AI and Genetics Join Forces to Map the Molecular Roots of Aging — and Find Drugs That May Already Treat Them
A new framework scans 12 age-related diseases simultaneously, uncovers 45 therapeutic targets, and delivers a striking finding: some of the most promising aging-related drug targets are already treatable with existing medicines.
Highlights
- New research proposes an AI-guided pipeline that combines pattern recognition with a genetic analysis technique, aiming to move from association to causation in aging target discovery.
- It identifies 45 aging-associated target genes across 12 age-related diseases.
- The most actionable implication is drug repurposing: several of the highlighted targets already have approved or investigational drugs, which could shorten the path from aging biology to clinical intervention.
One of the biggest frustrations in aging research is the gap between biological insight and clinical intervention. Scientists have catalogued the hallmarks of aging in rich molecular detail, yet translating that knowledge into actual drugs remains slow, expensive, and prone to failure. A new study published in Aging & Disease from researchers at Insilico Medicine proposes a way to dramatically accelerate that process by combining the pattern-recognition power of artificial intelligence with the causal rigour of human genetics.
The result is a target discovery pipeline that not only identifies genes associated with aging but also provides evidence that those genes are causally involved, and flags which ones are already targetable with approved or investigational drugs.
The Core Problem: Association Is Not Causation
For decades, genomic studies have generated long lists of genes correlated with aging and age-related diseases. But correlation is not causation, and the drug development pipeline is filled with targets that seemed biologically plausible but failed in clinical trials. One major reason is that most computational approaches identify genes that change during aging without establishing whether those changes actually drive disease or are merely passengers along for the ride.
The Insilico researchers set out to address this problem head-on, building a multi-layered framework that uses AI to sift through enormous multi-omic datasets encompassing genomic, transcriptomic, and proteomic information. All the while, it simultaneously applies a genetic technique to test for causality.
Twelve Diseases, Four Disease Areas, One Framework
The researchers developed an AI-driven target discovery framework that integrates large-scale datasets to prioritize therapeutic targets shared between aging and 12 age-related diseases. These diseases span across four major disease areas: neurological, inflammatory, metabolic, and fibrotic disorders (conditions of age-related tissue scarring).
This cross-disease design is deliberate and significant. Rather than studying Alzheimer’s disease, type 2 diabetes, or lung fibrosis in isolation, the framework explicitly searches for targets implicated across multiple conditions simultaneously. The logic is compelling: if aging itself is the primary driver of these diseases, then targets shared across many of them are more likely to reflect fundamental aging biology, and drugs that hit those targets could theoretically address multiple conditions at once.
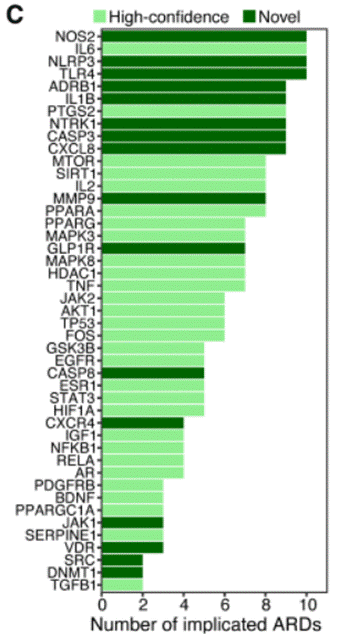
The researchers identified 29 high-confidence and 16 previously unrecognized aging-associated targets implicated across the selected disease areas. Together, these 45 targets represent a prioritized shortlist from what would otherwise be an unmanageable landscape of thousands of candidate genes.
Inflammation at the Center of Everything
When the team examined which biological pathways were consistently perturbed across aging and all twelve diseases, a clear pattern emerged. Convergent pathway perturbations were characterized by robust upregulation of interferon (a protein that helps coordinate immune defenses) and inflammatory signalling, alongside coordinated downregulation of cell proliferation signalling, consistent with heightened inflammatory activation and reduced tissue growth activity during aging.
In plain terms: as we age, the immune system becomes chronically dysregulated, while the cellular machinery responsible for growth and tissue renewal progressively winds down. Both shifts appear not only in aging itself but consistently across neurological, inflammatory, metabolic, and fibrotic conditions studied. Moreover, an assessment based on how the cellular pathways examined relate to the hallmarks of aging revealed chronic inflammation as the most enriched hallmark across aging and age-related diseases.
This convergence on inflammation is not entirely surprising; it aligns with a large and growing body of evidence. However, the scale and consistency of the signal here, detected independently across twelve distinct diseases using AI-driven analysis, adds substantial weight to the case that targeting inflammation is not just one strategy among many for aging research, but arguably the central one.
The Genetic Proof: Mendelian Randomization
The most methodologically important contribution of the study may be its use of Mendelian randomization. Mendelian randomization is a technique that exploits naturally occurring genetic variation to test for causal relationships, rather than merely statistical associations.
The principle works like a natural experiment. People are born with different genetic variants that, in effect, randomize them from birth into groups with slightly higher or lower expression of particular genes. Because these variants are assigned at random during conception (independent of lifestyle, diet, or environment), any association between a variant and a health outcome is much harder to explain away as something that could be explained by other factors, which would make the association weaker or false. It is the closest thing human observational research has to a randomized controlled trial, conducted across millions of people and thousands of generations.
Mendelian randomization provided genetic causal support for the genes IL6, IL6R, NLRP3, NOS2, TLR4, and GLP1R in aging-related traits and multiple age-related diseases. This finding highlights potential opportunities for drug repurposing. Accordingly, each of these six genes is a significant finding in its own right:
IL6 and IL6R encode interleukin-6 and its receptor, one of the master regulators of inflammatory signalling in the body. Drugs blocking this pathway (such as tocilizumab, originally approved for rheumatoid arthritis) already exist and are widely used in the clinic. The finding that IL6R has causal genetic ties to aging and multiple age-related diseases raises the prospect that such drugs could be repurposed for broader anti-aging applications.
NLRP3 encodes the master switch of the inflammasome—a molecular complex that triggers a particularly inflammatory form of cell death and has been strongly implicated in age-related conditions, including gout, cardiovascular disease, and neurodegeneration. Multiple NLRP3 inhibitors are currently in clinical development, and the new causal genetic evidence strengthens the case for testing them in aging contexts.
NOS2 and TLR4 encode key mediators of immune responses and nitric oxide signalling, both of which shift significantly with age and chronic disease.
GLP1R encodes the receptor targeted by the wildly successful GLP-1 drugs like semaglutide. It appears here as causally relevant not only to metabolic disease but to aging-related traits more broadly. This adds yet another layer to the expanding story of GLP-1 biology, which increasingly appears to have effects well beyond glucose control and weight loss.
A Genetic Signal Connecting IL6R to Human Lifespan
Perhaps the most striking individual finding involves a genetic analysis (known as a co-localization analysis) of the IL6R gene locus. This analysis demonstrated a shared genetic signal at the IL6R locus between gene expression levels and parental survival.
This means that the same stretch of DNA that influences how much IL6R a person expresses also influences how long their parents lived, a widely used proxy for heritable longevity. The overlap is not a coincidental overlap between two separate signals; it is the same underlying genetic variation doing double duty.
The co-localization analysis is considered strong evidence that a gene is genuinely involved in a biological process, rather than merely lurking nearby in the genome. In practical terms, this analysis strengthens the hypothesis that modulating IL-6 signalling, a druggable pathway with approved medications, could influence not just individual diseases but the pace of aging itself.
A Scalable Blueprint for Future Discovery
Beyond the specific targets identified, the broader contribution of the paper is the framework itself. The authors describe a pipeline that can, in principle, be applied to any set of diseases, expanded to include new data types, and updated as datasets grow. The findings outline a scalable AI-guided framework for identifying causal and repurposable therapeutic targets for aging and age-related diseases.
This scalability matters enormously. Drug development timelines are measured in decades; the pace at which aging biology generates new candidate targets has accelerated faster than the capacity to test them. Frameworks that can rapidly prioritize, filter, and validate targets computationally (before a single animal study is run) could meaningfully compress that timeline.
The drug repurposing angle is particularly attractive from a practical standpoint. Repurposed drugs already have established safety profiles, manufacturing processes, and in many cases regulatory approval pathways. If the causal evidence for targets like IL6R and NLRP3 proteins holds up in prospective trials, the path to clinical application would be far shorter than for an entirely novel therapeutic.
Caveats and Context
The study’s findings should be interpreted with appropriate caution. The framework is computational, and while Mendelian randomization is a powerful tool, it has its own limitations, including sensitivity to the quality of genetic instruments and the possibility of horizontal pleiotropy (where a genetic variant affects the outcome through pathways other than the intended target). The 45 targets identified represent prioritized candidates, not validated drugs; substantial experimental and clinical work lies ahead before any of them could be considered proven aging interventions.
It is also worth noting that the study comes from Insilico Medicine, a commercial AI drug discovery company with a natural interest in demonstrating the value of its platform. The researchers are transparent about this, and the methodology is scientifically sound, but independent replication and validation will be important.
Why This Matters for Aging Research
The study arrives at a moment of genuine momentum in the aging field. The conceptual shift toward treating aging itself as a disease, rather than accepting it as an inevitable background condition, has gained ground in both scientific and regulatory circles. The identification of causal, druggable targets shared across multiple age-related diseases feeds directly into that agenda.
If chronic inflammation, as the research suggests, is the most fundamental shared driver of aging and its associated diseases, then the field may already have the pharmacological tools needed to start intervening. The IL-6 pathway drugs exist. NLRP3 inhibitors are advancing through trials. GLP-1 receptor agonists are already transforming metabolic medicine and are being studied in a widening range of age-related conditions.
What has been missing is the systematic, causally grounded evidence to justify deploying these tools against aging itself, not just the individual diseases that aging causes. This study is a step toward supplying that evidence.
 Comments
Comments