Boosting NAD+ Levels Improves Heart Function in Mice with Heart Failure
A study from Duke University indicated that NMN supplementation improves function in mice with heart disease via boosting NAD+ levels and SIRT3 activity.
Approximately 6.5 million people in the United States had heart failure in 2019, and heart failure contributed to 1 in 8 deaths in 2017. The condition cost the nation an estimated $30.7 billion in 2012. In 2017, scientists from Duke University found that a molecule called nicotinamide mononucleotide (NMN) had therapeutic efficacy in mice with heart failure. Previous studies have shown supplementing with NMN improved heart function in multiple mouse models of heart failure, but the cellular mechanism with which this occurs remained poorly understood. The study provided a potential intervention strategy for patients with heart failure.
NMN Improves Heart Function to Stave Off Heart Failure
NMN increases the levels of nicotinamide adenine dinucleotide (NAD+), an essential molecule that influences various aspects of metabolism in the cell’s powerhouse, the mitochondria. Researchers have proposed that increased NAD+ levels with NMN supplementation enhance cardiac function via the activation of the protein SIRT3, an NAD+-dependent protein that participates in regulating cellular health.
Another important function of SIRT3 is to lower the level of molecular modifications of proteins, called hyperacetylation, in the mitochondria. However, the researchers observed an elevated level of molecular modification in genetically modified mice with heart disease, indicating that the animals have a reduced function in SIRT3. At five weeks of age in diseased mice, the researchers found no differences in the abundance of hyperacetylation when compared to normal mice. But as the heart disease progressed, at eight and 13 weeks of age, dramatic elevations in hyperacetylation were seen in mitochondrial proteins, suggesting a lack of SIRT3 functionality in the heart-diseased mice.
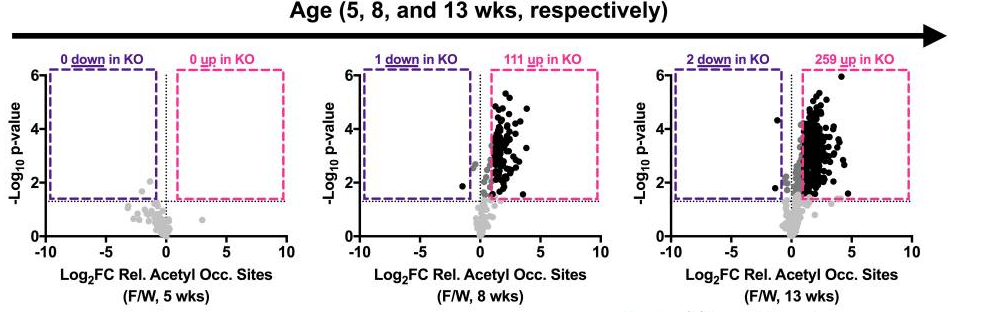
Moreover, ablating the gene for SIRT3, thereby getting rid of the protein, induced a cardiac dysfunction called ventricular wall thickening, demonstrating the pivotal role of SIRT3 in proper cardiac function. As the thickened walls become stiff, the amount of blood taken in and pumped out to the body reduces with each heartbeat, and the condition may lead to heart failure and in rare cases, even cardiac arrest.
Although the loss of function in SIRT3 may result in serious consequences, the team of researchers found that injecting NMN into mice with heart failure reduced hyperacetylation and improved cardiac function. The results indicated that NMN supplementation improves SIRT3 function. Deleting the SIRT3 gene to get rid of SIRT3 protein’s function resulted in the loss of NMN’s beneficial effects in the diseased animals.These observations provided further evidence that the effects of NMN depend upon SIRT3 in the heart.
The study showed that NAD+ therapy with NMN improves cardiac metabolic function and energy metabolism with SIRT3 playing a key role in mediating the effects. “In summary, our findings provide mechanistic insight into SIRT3-dependnet therapeutic effects of NMN treatment,” stated the scientist with reference to NMN’s effects on the genetically-modified mice with heart failure. The researchers also noted that NMN supplementation or SIRT3-activating therapy could benefit patients with cardiac dysfunction in the future.
 Comments
Comments