Human Cells Exposed to an NAD+ Precursor Have Different Survival Responses
Dihydronicotinamide riboside (NRH) promoted cell toxicity in cancerous human cells
Highlights
- Dihydronicotinamide riboside (NRH) exposure results in different toxic responses between cultured human cells.
- NAD+ increases can cause cellular dysfunction and eventually induce cell death in certain cell types.
Nicotinamide adenine dinucleotide (NAD+) is involved in processes that are intimately implicated in cell metabolism and aging. Our bodies are dependent on stable levels of NAD+, as decreases in NAD+ levels within cells are tied to multiple metabolic complications and age-related disorders like cardiovascular and Alzheimer’s disease. One way to keep NAD+ levels up is by supplementing with NAD+ precursors like dihydronicotinamide riboside (NRH), which greatly increases NAD+ levels in both cultured cells and mice. But whether the effects of NRH translate to human cells has not been fully elucidated.
Sonavane and colleagues from the University of South Alabama College of Medicine published an article in the journal PLoS One demonstrating cell type-specific effects of acute NRH exposure in human cells. NRH exposure resulted in different toxic responses between cultured human embryonic kidney cells and human liver cancer cells. Interestingly, NRH exposure led to poor cell survival of the cancer cells whereas the embryonic cells were unaffected in terms of their viability. This shows that NRH has specific effects in different tissue types as well as diseased versus healthy cells, which may have implications for how and by whom NRH is consumed.
Finding the right NAD+ precursor for human consumption
Studies have demonstrated that NAD+ levels decline with age in various tissues of rodents and humans. Critically, the decrease in NAD+ levels within cells is also known to cause multiple metabolic complications and age-related diseases, such as obesity, diabetes, and Alzheimer’s disease. Supplementation of NAD+ precursors such as nicotinamide mononucleotide (NMN), nicotinamide riboside (NR), nicotinic acid (NA), and nicotinamide (NAM) can replete the NAD+ pool. Yet, not all supplements share the same efficacy in mitigating metabolic complications including insulin sensitivity, fatty liver, and kidney damage, and preventing mitochondrial disease, Alzheimer’s disease, cardiovascular disorders, and even cancer.
To achieve protective effects of disease states, animal models require high doses (250 to 1000 mg/kg/day) of NAD+ precursors. These dose ranges correspond to a non-sustainable 18.5 to 75 grams per day supplementation for an adult, limiting the ability to pursue protective effects in humans based on the animal model work. In order to use these compounds in humans, we need to find the optimal NAD+ levels in human tissues and how to dose the NAD+ precursors accordingly to reach these levels. On top of that, the effectiveness and duration with which a precursor boosts NAD+ levels in these tissues also need to be understood.
Recent investigations have demonstrated that NRH increased NAD+ levels more significantly in both cultured cells and mouse models than current supplementation strategies with NR or vitamin B3 (nicotinamide and niacin). Since the consequences of extreme boosts in NAD+ levels are not fully understood, caution in the use of NRH must be exercised as it may have adverse effects. For these reasons, Sonavane and colleagues examined the cell-specific effects of acute NRH exposure in mammalian cells.
Not all human cells have the same reaction to NRH
The research team from the University of South Alabama College of Medicine looked at how different doses of NRH affected two human cell lines: one generated from embryonic kidneys (HEK293T) and another from liver cancer (HepG3). NRH exposure resulted in different cellular toxic responses between HEK293T and HepG3 cells. The liver cancer cells show dose-dependent toxicity when supplemented with NRH (100 to 1000 μM), but the human embryonic kidney cells were unaffected. The toxic effects observed in HepG3 but not in HEK293T after NRH exposure highlights the importance of NRH specific effects in different tissue types, such as diseased ones that are cancerous.
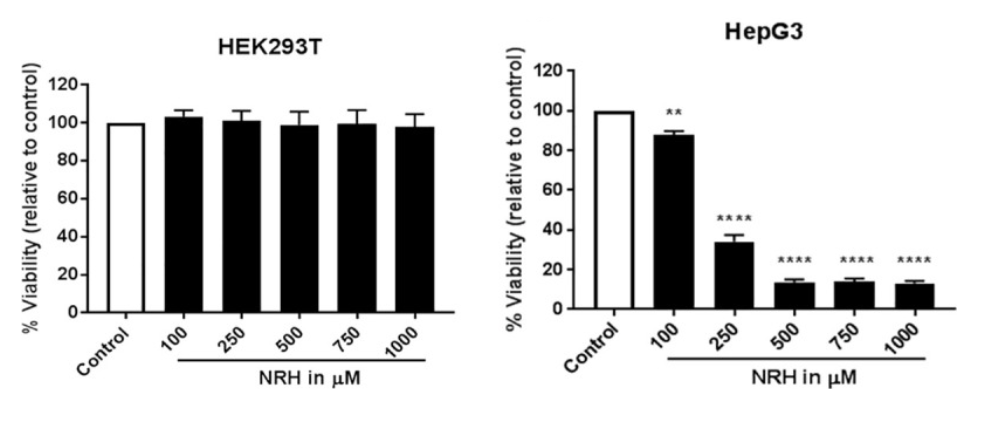
When supplementing HepG3 with a low amount of NRH (100 μM), Sonavane and colleagues observed a significant increase in reactive oxygen species (ROS) — oxygen-containing compounds that can damage cells and their DNA. Along these lines, NRH induced mitochondrial DNA damage in those cells and the levels of proteins that demarcate cell death (BAX and PUMA), which was linked to cell death triggered by the mitochondria. It is not surprising then that the research team saw functional changes in the HepG3 cells treated with NRH, such as metabolic dysregulation and mitochondrial dysfunction.

These observations suggest that cell-specific responses to NAD+ increases can cause an imbalance in NAD+ pools within different cell compartments, leading to cellular dysfunction and eventually inducing cell death. The cell-specific effects are likely mediated through the different metabolic fate of NRH in these cells, which warrants further study in other human cell types. These findings give us a better understanding of the cellular fate within specific tissues and cells of lower levels of NAD+ precursors, which is needed to create more beneficial supplements for boosting NAD+ biosynthesis and reduce its harmful side effects.
 Comments
Comments