Mutations Cause NAD+ Deficiency Linked Organ Malformation
Researchers propose that NAD+ precursors may aid future pregnancies for families with Congenital NAD Deficiency Disorder.
Highlights
· Mutations that induce impaired NAD+ synthesis facilitate heart, kidney, vertebral, and limb malformations in a condition called Congenital NAD Deficiency Disorder.
· This study expands the list of known disorder-associated mutations in enzymes participating in NAD+ synthesis.
· The researchers suggest that certain mutations can cause organ malformation due to reduced enzyme activity in NAD+ synthesis.
Congenital NAD Deficiency Disorder describes a condition where DNA mutations affect the levels of nicotinamide adenine dinucleotide (NAD+) — an essential compound for all cells — which are insufficient and affect human development. These patients have malformations in the heart, kidney, vertebrae, and limbs during development. The mutations underlying this disease have not all been accounted for, and we do not fully understand how they relate to the disorder’s manifestation and severity, which is crucial for its diagnosis and treatment.
Dunwoodie and colleagues from the University of New South Wales in Australia published a study in Human Mutation where they identified new mutations linked to Congenital NAD Deficiency Disorder. These mutations map to three genes involved in the NAD+ synthesis pathway that, when introduced to yeast, cause moderate to severe NAD deficiency analogous to insufficient synthesized NAD+ in patients. The findings help pinpoint where alterations in the NAD+ synthesis pathway occur, which may aid in future therapeutic development that target these affected enzymatic steps.
NAD+ Is Essential For Human Development
During gestation, all mammals, including humans, synthesize and use NAD+ — an essential cofactor involved in over 400 cellular reactions that is critical to human development. This NAD+ is synthesized from scratch using the protein building block L‐tryptophan obtained from the diet through a pathway called the kynurenine pathway.
It’s known that mutations in three genes — KYNU, HAAO, and NADSYN1 — encoding enzymes of the kynurenine pathway disrupt NAD+ synthesis and occur in patients with multiple malformations of the heart, kidney, vertebrae, and limbs. These Congenital NAD Deficiency Disorder patients have different DNA mutations that possibly affect human development in different ways. However, not all the DNA mutations that cause Congenital NAD Deficiency Disorder are known or how they affect human development.
Study Identifies Novel Mutations In NAD+ Biosynthesis Enzymes
To better understand the link between DNA mutations that perturb NAD+ synthesis and abnormal human development, Dunwoodie and colleagues looked at the mutations of seven patients from different families with developmental defects that are reflective of Congenital NAD Deficiency Disorder. They found that these patients had novel mutations in two of the genes encoding enzymes in the kynurenine pathway — 3-hydroxyanthranilate 3,4-dioxygenase (HAAO) and kynureninase (KYNU).
Three patients had mutations in the enzyme HAAO, which is involved in making NAD+ from the protein building block L-tryptophan. All three patients with HAAO mutations exhibited defects consistent with previously published patients: malformations affecting the heart, vertebrae, limbs, and kidneys.
Dunwoodie and colleagues also examined four patients with gene mutations for the enzyme kynureninase (KYNU), which is also involved in synthesizing NAD+ from tryptophan. These individuals exhibited limb defects, heart abnormalities, and altered facial features like wide-spaced eyes, a short neck, and a broad nose — all hallmarks of Congenital NAD Deficiency Disorder.
Not All Mutations Are Created Equal
Dunwoodie and colleagues then used computer algorithms to predict the severity of these DNA mutations to the function of the protein they encode. These programs predicted that these DNA mutations had high pathogenicity — the property of causing disease.
To verify if these mutations affected NAD+ synthesis, the research team inserted the mutations into yeast, which share the same NAD+ synthesis pathway enzymes as humans. By monitoring the development and NAD+ production of these yeast, Dunwoodie and colleagues inferred whether these mutations can affect NAD+ synthesis in humans. When they generated yeast carrying these mutations in HAAO, the single-celled organisms displayed significantly reduced growth and decreased total NAD+ levels. Similarly, when they inserted the KYNU mutations from these patients into yeast, they saw reductions of at least 57% compared to that of healthy, non-mutated yeast.

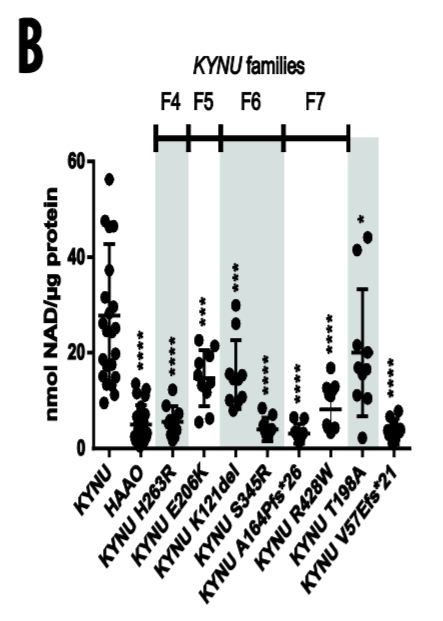
To date, all HAAO or KYNU mutations identified as causal of Congenital NAD Deficiency Disorder result in the complete loss of function of these enzymes. In this study, the Australian research team identified multiple rare damaging mutations in HAAO or KYNU that result in moderate to complete loss of function and NAD deficiency. Generally, the more severely compromised NAD synthesis in yeast caused by these variants, the greater the number of malformations observed in affected patients.
Vitamin B3 Supplementation May Reduce Pregnancy Outcome Severity
This study adds to our understanding of Congenital NAD Deficiency Disorder by expanding the spectrum of defects caused by embryonic NAD deficiency and establishes that rare, damaging mutations within HAAO or KYNU can cause malformation due to reduced enzyme activity. This suggests that there can be combinations of more common and less damaging gene mutations that manifest a range of traits associated with NAD deficiency.
“We have identified seven new cases of Congenital NAD Deficiency Disorder in humans stemming from rare biallelic variants in [3-hydroxyanthranilate 3,4-dioxygenase] or [kynureninase], exhibiting multiple malformations consistent with insufficient NAD during embryogenesis,” stated Dunwoodie and colleagues in their publication.
How embryo developmental processes (embryogenesis) get disrupted by NAD+ deficiency remains unclear. But, Dunwoodie and colleagues say that boosting mothers’ NAD+ levels during pregnancy with vitamin B3 precursor supplementation might reduce babies’ developmental abnormalities from NAD+ deficiency.
The study also showed that the Congenital NAD Deficiency caused by these newly-identified mutations results from the lost function of key NAD+ synthesis enzymes. This helps us to understand that this disorder may often arise from diminished or lost NAD+ synthesis enzyme function. Researchers can then use this knowledge to develop therapeutic options that restore the action of these enzymes.
 Comments
Comments