NAD+ Boosting Protects Human Cells From Premature Aging Disease
Researchers fight off childhood neurodegenerative disease characteristics rooted in NAD+ deficiency, mitochondrial damage, and senescence
Highlights
· Mitochondrial dysfunction drives premature aging seen in ataxia telangiectasia (A-T).
· Enhancing mitochondrial recycling by boosting NAD+ is a potential therapeutic intervention for (A-T).
Ataxia telangiectasia (A-T) is a devastating, complex genetic disorder characterized by degeneration of the nervous system often during infancy or early childhood. Premature aging diseases like A-T are often linked to breakdown and leakage of the battery packs that generate energy for our cells (mitochondria) and senescence — an arrest in cell growth and replication that’s necessary to keep our organs from decaying. These phenomena happen in natural aging too, but don’t take off so early in life and at such a rapid rate. But whether cells freeze up and mitochondria crash hasn’t been explored regarding A-T.
In an article published in Aging Cell, Yang and colleagues from the National Institute on Aging demonstrate that the buildup of damaged mitochondria and senescent cells occurs in cells from A-T patient as well as cultured human cells and mice that model the disease. The research team based out of Bethesda, Maryland, find that boosting levels of nicotinamide adenine dinucleotide (NAD+) — a molecule at the core of many processes, including mitochondrial function and recycling — clears damaged mitochondria and prevents senescence in A-T models.
These findings link the neurological symptoms of A-T directly to senescence and the loss of healthy mitochondrial populations within cells.
“Our data support the concept that targeting the maintenance of mitochondrial quality may have potential roles in the prevention of senescence and neuroinflammation in neurodegenerative diseases,” concluded Yang and colleagues.
Rogue DNA Starts Cascade Towards Neurodegeneration Linked to A-T
In humans, loss of a particular molecular machine (enzyme) results in A-T, a rare inherited genetic disease characterized by neurodegeneration as well as cancer predisposition, sterility, and immune deficiency. A-T patients also suffer from a variety of inflammatory characteristics, which are thought to be rooted in the failed development of certain immune cells.
The enzyme ATM kinase, which is encoded by the ataxia telangiectasia-mutated (ATM) gene, is a master regulator of the DNA repair responses. When DNA gets damaged, ATM gets activated. While some major features of A-T reflect inefficient DNA repair, how this all translates into neurodegeneration in A-T is poorly understood.
There are clues that unresolved DNA damage can impair mitochondrial function, promote disease development, and accelerate aging, as reported in A-T. One clue about the inner workings of A-T may lie in the growing evidence that persistent DNA damage and senescence are linked.
Malfunctioning Mitochondria Pave the Way to Aging
Another driver of age-related decline is the loss of mitochondrial function. Multiple lines of evidence point to mitochondrial dysfunction as a component of A-T features. Dysfunctional mitochondria can induce senescence in cultured cells and animals. Driving mitochondrial function is a molecule called NAD+, which is deficient in ATM-deficient neurons. But, little is known about the connections between senescence, inflammation of the nervous system, mitochondrial dysfunction, and NAD+.
DNA Leaking from Mitochondria Links to Senescence in A-T
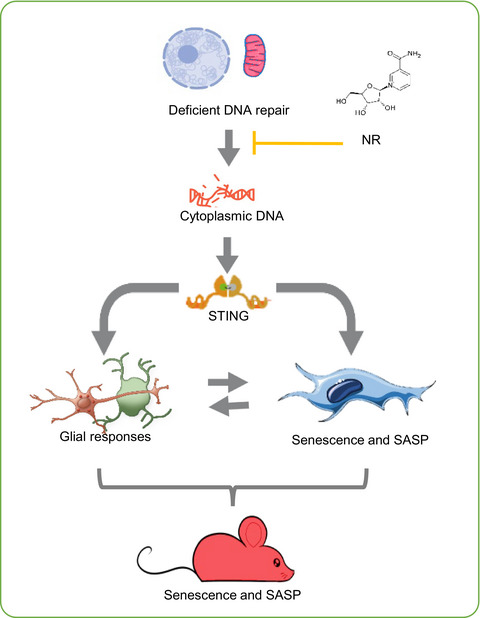
In this study, Yang and colleagues looked at whether mitochondrial dysfunction and senescence were at play in cells from A-T patients as well as ATM-deficient neural cells and mice. They found that cells from A-T patients and those lacking ATM have impaired mitophagy — the process of clearing damaged mitochondria — and consequently promotes the release of mitochondrial DNA into the cytoplasm. This build up of cytoplasmic DNA triggers an antiviral immune response in the brain called STING, which initiates a robust pro-inflammatory response and senescence linked to the deficient health span in ATM-deficient mice.
Enhanced NAD+ Equilibrates Mitochondrial Populations
Yang and colleagues went on to show that the accumulation of fragmented DNA floating around in the cytoplasm dropped by boosting the cell levels of NAD+. They think that the NAD+ boosting works by activating mitophagy because the NAD+ precursor nicotinamide riboside (NR) failed to prevent senescence following inhibition of mitophagy.
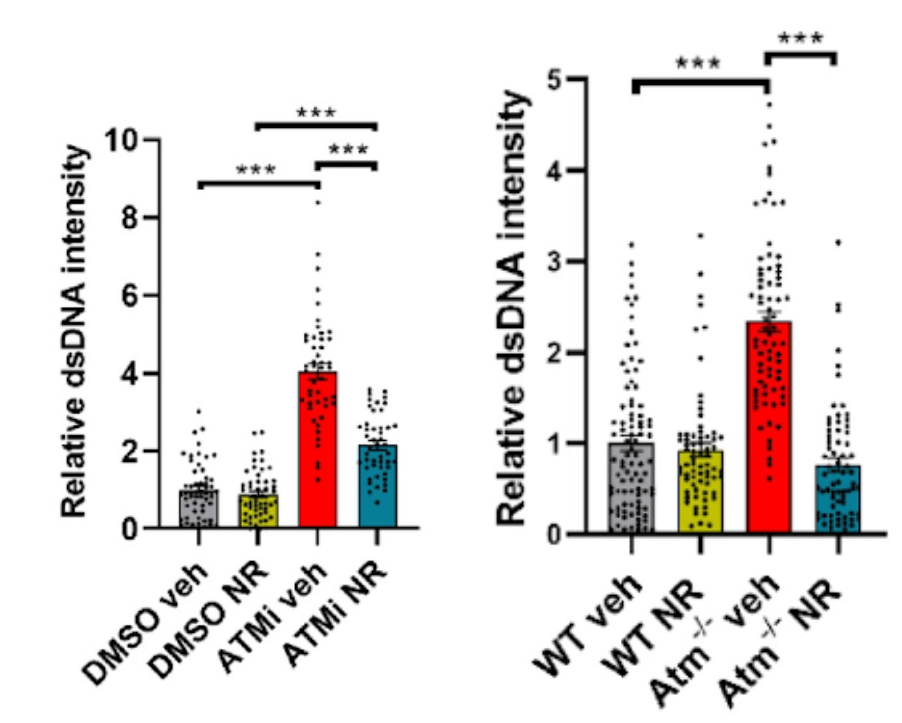
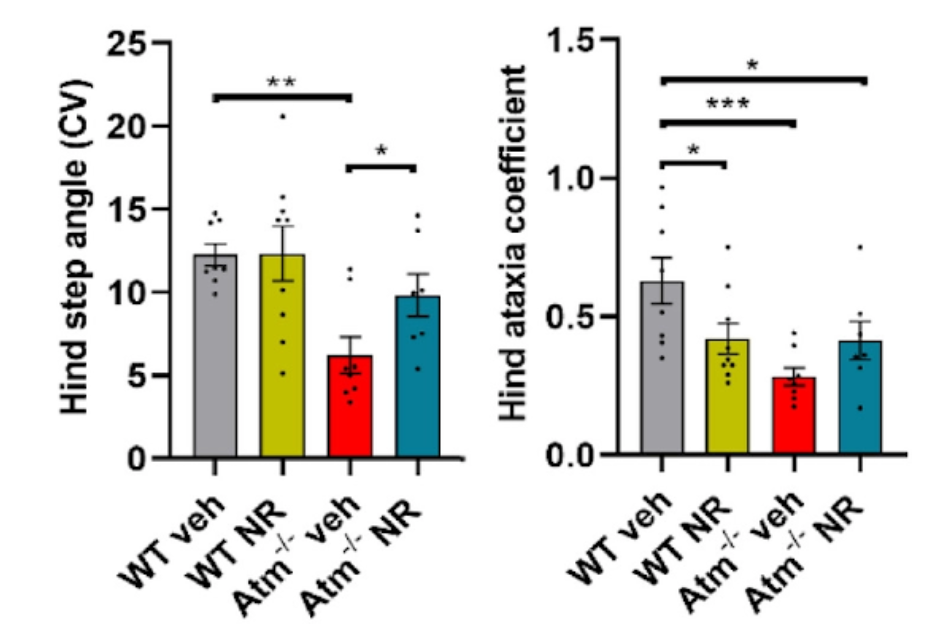
The National Institute on Aging research team also found that the effects of enhancing NAD+ levels affected not only ATM-deficient cells but also at the level of behavior. In mice lacking ATM, NR prevented neuroinflammation and senescence through enhancing mitochondrial function, reducing cytoplasmic DNA and preventing activation of STING in ATM-deficient cells and mice. ATM-deficient mice also regained motor function when their NAD+ levels were restored.
Can NAD+ Boosting Push Back Aging?
As NAD+ plays important roles in a multitude of molecular and cell processes, including DNA repair, mitochondrial function, and senescence, supplementation of NAD+ is critical and beneficial in settings like A-T or normal aging where NAD+ levels are low. But whether elevating NAD+ levels can prevent aging in humans remains to be determined. Studies are needed to test whether this is applicable in A-T patients and other premature aging conditions prior to be made available in a clinical setting.
 Comments
Comments