NAD+ Increases Healthspan and Lifespan Through DNA Repair in Animals with Neurological Defects
An international team of scientists from Europe, the US and Australia revealed that boosting NAD+ mitigates DNA damage and improves survival in animals with neurological defects in a 2016 study.
Ataxia telangiectasia (A-T) is a rare genetic disease consisting of uncoordinated movement, sensitivity to radiation, and atrophy of a region of the brain called the cerebellum. The disease presents in individuals who inherit a pair of defective genes, one from each parent.
The gene Ataxia-Telangiectasia Mutated (ATM) regulates cell growth and division under normal circumstances, but defects in it lead to abnormal cell death and cause A-T. Between 0.5% and 1% of the general human population has one copy of the disease-causing form of this gene, resulting in an elevated risk of cancer and age-related diseases. Currently, there is neither a cure nor a specific intervention for the patients and carriers with the faulty ATM gene.
Patients with A-T have low cellular levels of nicotinamide adenine dinucleotide (NAD+). NAD+ constitutes a key cellular molecule in metabolism, stem cell rejuvenation, neuroprotection, and longevity. No one had investigated the effects of replenishing NAD+ in animals with A-T until 2016.
NAD+ Restoration Improves Neurological Health
A team of international scientists from Europe, the US and Australia found restoration of NAD+ reduced the severity of neurological defects and improved the healthspan and lifespan of these diseased animals. Their study published in Cell Metabolism indicates that the beneficial effects come from NAD+ stimulating DNA repair and the health of the mitochondria, the cell’s powerhouse.
The team studied animals with deleted or reduced expression of ATM gene, including neurons from rats, along with mice and roundworms. Their study revealed replenishing NAD+ with the NAD+ booster, nicotinamide riboside (NR), improved the signaling and overall health of mitochondria in rat neurons with reduced expression of the ATM gene. NR treatment of neurons with reduced ATM gene activity reduced DNA damage and cell death, known as apoptosis.
The scientists also studied roundworms with reduced ATM gene activity and discovered that NR treatment promotes DNA repair. The experimentation revealed exposing these worms to ionizing radiation reduced the number of offspring of the worms by 40%. But NR treatment improved survival of the radiation-exposed worms from 41% to 79.6% during a stage of development, the late embryonic stage. During this stage, only a particular DNA repair pathway, the nonhomologous end-joining pathway, can repair DNA breaks from radiation. The scientists thought these results demonstrated NR treatment promotes DNA repair from ionizing radiation.
In mice without the ATM gene, NAD+ supplementation improves healthspan in the animals. Mice with deleted ATM gene had 40% less NAD+ in the region of the brain called the cerebellum compared to normal mice. However, after NR treatment in drinking water, their NAD+ levels returned to approximately normal levels. Moreover, the ATM-absent mice only performed half as good normal mice in the maze test that assess their cognitive function. But after NR treatment they performed as well as normal mice, showing an improvement in cognitive function, a key aspect of healthspan. ATM-deleted mice treated with NR had a significantly extended lifespan as well. Although animals without the ATM gene die before they reach 150 days old, approximately 80% of those that received NR treatment live past 150 days.
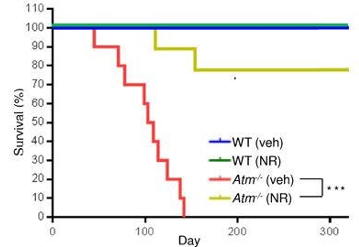
(Fang et al., 2016 | Cell Metabolism) The figure illustrates the ATM knockout mice without NR supplementation have zero survival around day 150 after birth. ATM knockout mice supplemented with NR had around 80% survival at 150 days of age. These results indicate NR drastically improves survival in Atm knockout mice.
“In summary, our results suggest that NAD+ depletion can induce severe neurodegeneration,” stated the authors. “While it remains to be seen how well NAD+ supplementation translates to the clinical treatment of A-T patients, our findings suggest novel therapeutic strategies to combat this disease, and possibly other premature aging disorders with DNA repair defects.” Administering supplements boosting NAD+ levels could provide a therapeutic option for the treatment of these neurological defects.
 Comments
Comments