NAD+ Levels Delay Facial Nerve Degeneration After Trauma
Compounds modulating NAD+ metabolism are new therapeutic candidates in facial nerve injury
Highlights
- Following facial nerve injury, neurodegeneration and neuroinflammation are suppressed in mice lacking an enzyme that depletes levels of the bioenergetic compound NAD+.
- Supplementation of an NAD+ precursor slowed the nerve projection degeneration in mice with facial nerve injury.
Facial muscles make all kinds of motions, often to convey emotion. The nerves that control these muscles are what allow you to raise your eyebrows, frown, smile, and pout. When these nerves get damaged, these facial movements go away, and they rarely recover as they don’t regenerate on their own or with treatment. So, there is a real need for figuring out how to restore these nerves to bring the faces of people suffering from facial paralysis back to life.
In an article published in Scientific Reports, Takaso and colleagues from Kanazawa University analyzed the role of a candidate enzyme for regulating neurodegeneration and neuroinflammation called CD38 on a model of facial paralysis in mice. Mice lacking CD38 that underwent a facial nerve axotomy — the severing of nerve cell projections in the face — showed delayed degeneration of the nerve projections called axons. When they supplemented levels of NAD+, the molecule that CD38 breaks down, they also saw slower axon degeneration.
This study suggests that CD38 depletion and NAD+ supplementation may protect severed axons in facial paralysis. “Our result may raise the possibility of the treatment for facial nerve axotomy from the view of suppressing nerve degeneration by focusing on NAD+ metabolism,” proposed Takaso and colleagues.
What Happens During a Facial Nerve Injury Response?
Facial nerve axotomy is an injury that occurs typically in trauma and upon surgery. Clinically, the cure rate is low even after nerve reconstruction, and patients often have sustained facial paralysis and involuntary tremors. Following facial nerve axotomy, both the cell body, which lies in an area called the facial nucleus, and the axon degenerate.
During the processes after facial nerve axotomy, the surrounding environment is also dramatically changed, and has an impact on the fate of the transected nerve; supporting nerve cells and immune-related cells called astrocytes and microglia, respectively, are activated in the facial nucleus, and immune cells called macrophages infiltrate the transected nerve. These cells can produce proinflammatory molecules that cause nerve damage.
NAD+ Metabolism Is Linked to Nerve Health
CD38 is a membrane-associated enzyme that consumes nicotinamide adenine dinucleotide (NAD+) — an essential compound for a myriad of cellular processes including metabolism. Accumulating evidence suggests that the deletion of CD38, which protects levels of NAD+, reduces neurodegeneration and neuroinflammation.
Furthermore, administration of nicotinamide riboside (NR), an NAD+ precursor, has been shown to prevent degeneration of certain nerve cell projections in the brain. These results suggest that CD38 depletion and high levels of NAD+ in the brain suppress neurodegeneration and neuroinflammation. But the neuroprotective effects of NAD+ and CD38 on facial nerve damage remain to be clarified.
Levels of NAD+ Attenuate Facial Nerve Damage Response
In the current study, Takaso and colleagues investigated the effect of NAD+ levels in a mouse facial nerve axotomy model. To do so, they first looked at how deletion of CD38 affected the response to facial nerve injury. Mice lacking CD38 showed delayed axon degeneration and demyelination — when the protective coating of nerve cells called myelin experiences damage — following facial nerve axotomy. These CD38-deficient mice also had suppressed infiltration of inflammatory immune cells called macrophages and microglia at the transected nerve. These results suggest that CD38 deletion particularly affects axon degeneration, demyelination, and neuroinflammation at the local site of facial nerve damage.
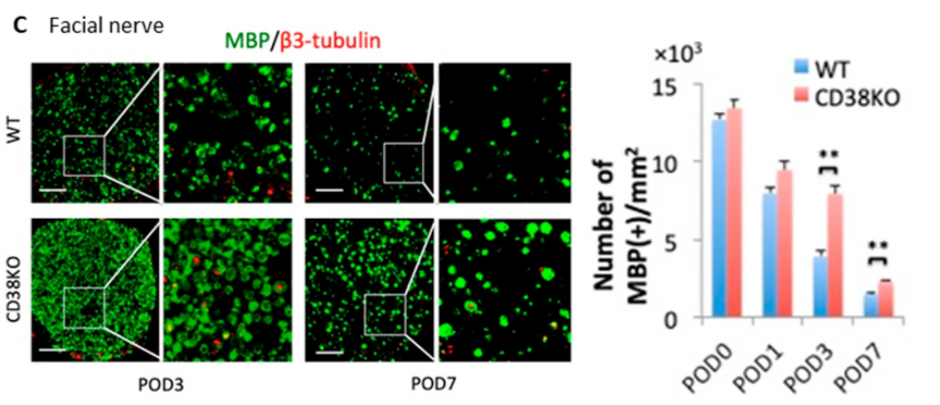
Takaso and colleagues then asked whether NAD+ boosting with NR had protective effects against axonal degeneration and demyelination in mice. Both axon degeneration and demyelination were delayed in mice treated with NR (400 mg/kg) from 3 to 7 days after axotomy. However, NR supplementation did not alter the level of facial nerve cell survival or macrophage infiltration after axotomy. “In facial nerves, our results suggest that the increase of NAD+ is more directly associated with preservation of the axon than suppression of immune cell infiltration,” proposed Takaso and colleagues.
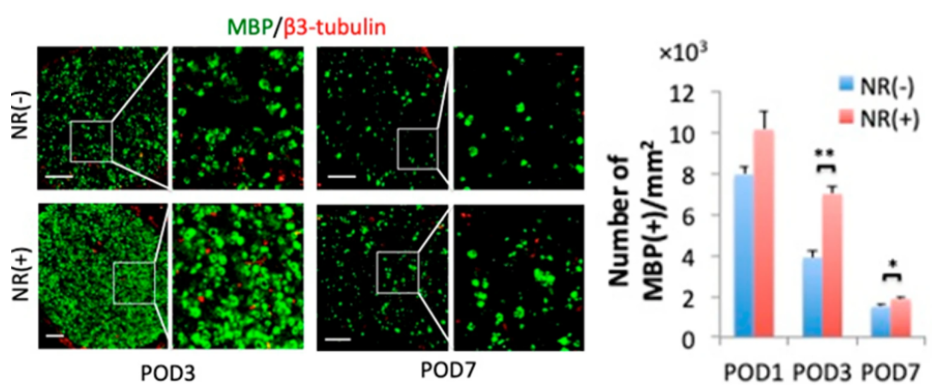
Given these findings, Takaso and colleagues propose that different compounds modulating NAD+ metabolism may serve as new therapeutic candidates for facial nerve injury. Combining NAD+ supplementation and CD38 inhibition with surgical treatment may become the new treatment to improve the outcome of facial nerve axotomy.
 Comments
Comments