NMN Protects Mouse Hearts from Failure
Study shows that NMN preserved mitochondrial health to protect the mouse heart from injury.
Approximately 647,000 people die from heart disease each year in the US, which equates to 1 in every 4 deaths. In the years 2014 and 2015, heart disease cost the United States about $219 billion each year, which includes costs from health care services, medicines, and lost productivity due to death. Finding therapeutic interventions in heart disease not only will improve the patients’ life quality but also lighten the economic burden on the society.
Scientists from Case Western Reserve University, who published a study in the Journal of Molecular and Cellular Cardiology, found nicotinamide mononucleotide (NMN) supplementation promotes the health of the cell’s powerhouse, the mitochondria, and prevents heart failure. Previous studies have associated mitochondrial dysfunction with heart failure, and researchers have recognized reductions in nicotinamide adenine dinucleotide (NAD+) levels in mitochondrial dysfunction.
Mice Hearts With Stressed Mitochondria Have Decreased NAD+ Levels
Many enzymes require NAD+ to function, including sirtuins. These proteins play essential roles in cell health and aging by removing molecular tags called acetyl groups from other proteins in the cell. The regulation of removing acetyl groups from proteins — a process called deacetylation — is critical to maintaining mitochondrial health. In the heart, the predominant sirtuin is SIRT3.
For the study, the researchers used mice with hearts deficient in a molecule called KLF4, which is critical for the mitochondrial health of heart cells. The mitochondrial proteins in mice hearts deficient for KLF4 had significantly higher amounts of acetyl groups, indicating less deacetylation and reduced SIRT3 function. These show that KLF4-deficiency leads to reduced deacetylation, or hyperacetylation, of mitochondrial proteins that predispose the mitochondria and heart to stress.
Zhang and colleagues then physically stressed out the hearts in these mice by blocking the aorta, which increases pressure on the heart and makes it work harder. This led to even greater reduced Sirt3 levels and lower NAD+ concentrations in KLF4-deficient hearts. In addition to being at low levels, SIRT3 function was likely decreased because of less available NAD+, reducing deacetylation of mitochondrial proteins and ultimately degrading mitochondrial health.
NMN Protects Mice with Mitochondrial Stress from Heart Failure
To raise the levels of NAD+ content in the mice, the researchers injected the animals with NMN, a NAD+-boosting molecule. NMN treatment improved the function of these hearts, increasing the amount it could pump under stressful conditions. It also prevented the amount of death of heart cells.
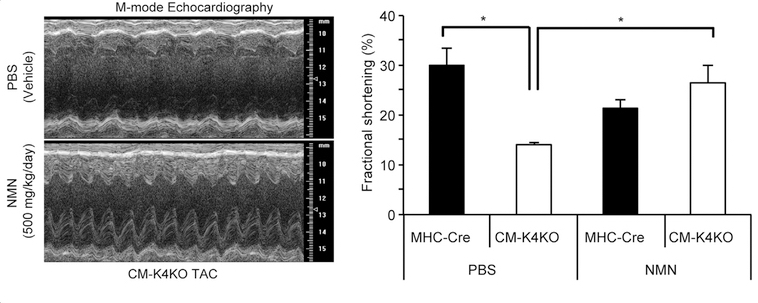
KLF4-deficient mice typically have high mortality rates, but NMN administration resulted in a 100% survival rate in these mice following the induction of high blood pressure in the heart. These results demonstrated NMN prevents heart failure in these mice sensitive to blood pressure-induced heart failure.
Zhang and colleagues found that the prevention of heart failure and mitochondrial health were correlated. Although KLF4-deficient mice typically have degenerated and fragmented mitochondria, NMN treatment almost completely restored the structure and function of the mitochondria.
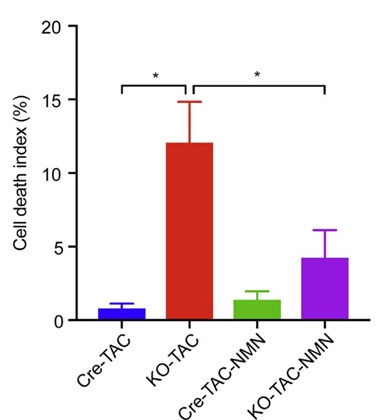
“Short-term administration of NMN, a precursor of NAD+, preserved mitochondrial homeostasis and rescued heart function from pressure overload-induced heart failure,” stated the scientists of the study. They go on to say that this study is proof that administration of NMN (even short-term) could be an effective therapy for heart failure, at least in mice.
 Comments
Comments