NMN Alleviates Lung Damage in Cigarette-Induced Pulmonary Fibrosis
Nicotinamide mononucleotide (NMN) clears aged, non-proliferating lung cells from cigarette smoke.
Highlights
- NMN treatment restores damaged cell cleanup processes to prevent aged, non-proliferating cell buildup following cigarette smoke exposure.
- Alleviating the buildup of these cells may prevent the progression of the fatal condition pulmonary fibrosis.
We all know that smoking is bad for our health. It’s also a major contributing factor to the development of idiopathic pulmonary fibrosis (IPF) — a fatal, age-related lung disease that stems from lung tissue scarring. Little is known about how IPF develops, but we do know that cigarette smoke is a substantial contributing factor along with the buildup of aged, non-proliferating cells — what researchers call senescent cells. But there’s more to be learned mechanistically about how cigarette smoke may tie into senescent cell buildup and how to slow the age-related progression of IPF.
Zhang and colleagues from Southern Medical University in China published a study in Free Radical Biology and Medicine where they found nicotinamide mononucleotide (NMN) treatment stimulates damaged cell cleaning – autophagy – of senescent cells in mice exposed to cigarette smoke. The research team found a pathway through which cigarette smoke drives the buildup of harmful, oxygen-containing molecules called reactive oxygen species (ROS) in the cell’s power-generating structure, the mitochondria, to facilitate senescent cell buildup. This mitochondrial ROS could be removed by autophagy stimulated by NMN to alleviate cell senescence and subsequent IPF. If these results translate to humans, NMN may provide a means to stave off IPF, especially in adults who smoke.
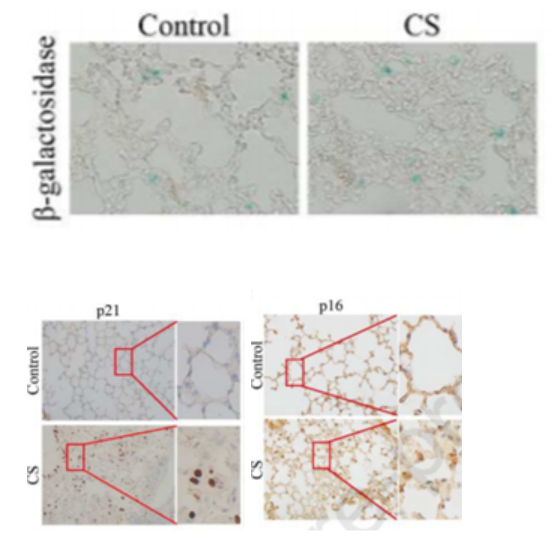
Cigarette Smoke Induces Lung Cell Senescence
To test the effects of cigarette smoke on cell senescence, Zhang and colleagues exposed mice in a chamber to cigarette smoke from five cigarettes for two 30-minute sessions daily for four weeks. In doing so, they found that cigarette smoke induced lung cell senescence. They also found increased senescence in lung cells treated with a cigarette smoke-infused extract (CSE) in laboratory dishes. These results strongly suggest that cigarette smoke induces lung cell senescence.
Impaired Autophagy Induces Lung Senescent Cell Buildup
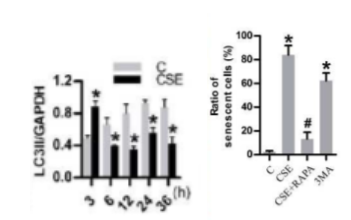
Previous studies by Zhang and colleagues indicated that impaired autophagy contributes to cigarette smoke-induced lung fibrosis. So, the team tried to link impaired autophagy to cigarette smoke-induced cellular senescence since the role of autophagy in senescence remained murky. They found that a protein related to autophagy called LC3 II initially increased but then dropped in lung cells cultured in laboratory dishes following their treatment with a cigarette smoke-infused extract (CSE). Moreover, they used a pharmaceutical agent called rapamycin to induce autophagy, which prevented CSE-induced lung cell senescence. These results show that, although autophagy was induced initially following CSE exposure, CSE ultimately reduces autophagy to induce senescence.
Harmful Oxygen-Containing Molecules Contribute to Cell Senescence
Since the buildup of ROS in mitochondria has become a hot topic in aging and cell senescence, Zhang and colleagues wanted to know whether mitochondrial ROS buildup results from cigarette smoke-induced impaired autophagy. The researchers found elevated mitochondrial ROS levels in CSE-treated lung cells and that treatment with mitoquinone, a compound that clears ROS, protected cells against senescence. They concluded that impaired autophagy resulted in harmful mitochondrial ROS buildup resulting in senescence.
NMN Restores Autophagy to Mitigate Cell Senescence
NMN is a precursor to the vital molecule nicotinamide adenine dinucleotide (NAD+) that plays key roles in cell-energy generating reactions and binds to proteins called Sirtuin1 that depend on NAD+ to function. So, by increasing NAD+ levels, NMN promotes Sirtuin1 function to remove molecular tags called acetyl groups from other proteins to activate them.
The team of researchers replenished NAD+ levels with an NMN concentration of 500 µM to find out what effect it has on autophagy. NMN promoted autophagy and suppressed cell senescence in lung cells as shown by increased autophagy-related LC3 II levels following CSE exposure plus NMN and reduced cell senescence markers with NMN, respectively.
In the pulmonary fibrosis pathway that Zhang and colleagues uncovered, Sirtuin1 removes acetyl groups from the autophagy modulator protein LC3 II to induce autophagy, mitigate mitochondrial ROS, and clear senescent cell buildup. Treatment with the Sirtuin1 inhibitor Ex527 reversed NMN’s effects. NMN treatment may therefore offer a means to restore autophagy and mitigate cigarette smoke-induced cell senescence and subsequent IPF onset.

Will NMN’s Effects Translate to Human Disease?
“We confirmed that cigarette smoke inhibited SIRT1 activity,” stated Zhang and colleagues in their publication. “Furthermore, we proved that SIRT1 activator and supplementation of NAD+ with its precursor could restore SIRT1 activity and prevent [lung cell] senescence,” they said in reference to using a Sirtuin1 activator or NMN to prevent lung cell senescence.
This study by Zhang and colleagues sheds some light on how cigarette smoking can contribute to the onset of lung diseases like IPF. Researchers will still need to conduct future human clinical trials to determine whether the benefits of NMN in preventing senescent cell buildup and IPF apply to people who smoke.
 Comments
Comments