NMN as Potential Therapeutic Agent for Neuromuscular Disease
Scientists from the University of Missouri report disruptions of the NAD+ biosynthesis pathway in mice facilitating malformed mitochondria and cell structure in muscle. Furthermore, nicotinamide mononucleotide (NMN) administration at least partially restores these cellular defects.
Scientists from the University of Missouri report disruptions of the NAD+ biosynthesis pathway in mice facilitating malformed mitochondria and cell structure in muscle. Furthermore, nicotinamide mononucleotide (NMN) administration at least partially restores these cellular defects.
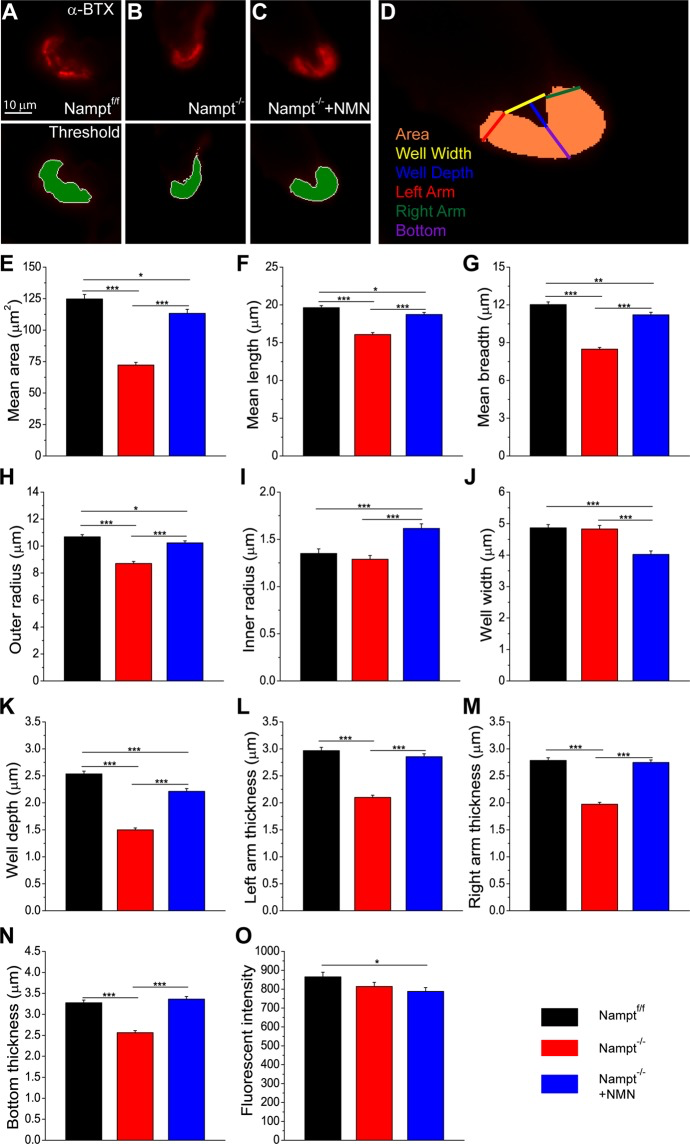
Image from Lundt et al. (2020)
Amyotrophic lateral sclerosis (ALS) constitutes a lethal motor neuron disease with degenerative changes in motor neurons.3,5 Onset of the disease typically occurs in the latter part of middle life. Symptoms of ALS include severe, progressive muscle atrophy and weakness, with complications of respiratory muscles leading to survival of two to four years following disease onset, typically.1,3 Treatment for ALS includes symptom management, respiratory support, and medications, which give modest benefits only in some patients.3,4,6 Previous research reveals ALS onset comes in large part from genetics (high heritability); however, identifying gene markers in order to generate treatment options remains elusive.3 Moreover, the complexity of ALS with varying symptoms and presentation of disease (phenotype) make it difficult to translate research using animal models of ALS to human clinical trials.3
Scientists propose many disease mechanisms in ALS, including disturbances in metabolism, dysfunctional proteins, mitochondrial dysfunction, and dysfunctional neurons. Studies need to clarify timing and degree to which each of these mechanisms contributes to disease progression (pathogenesis).3 Research continues on these mechanisms; and some believe involvement of nicotinamide adenine dinucleotide (NAD+) in mitochondrial, neuronal, and protein dysfunction plays a key role in ALS.
Lundt et al. (2020) perform a study looking at how disruptions in the NAD+ biosynthesis pathway affects the microscopic regions where nerves meet muscle (neuromuscular junctions) along with muscle function and microscopic structure of muscle in a mouse model. The group of scientists looks at muscle health at the cellular level with disruptions of the NAD+ biosynthesis pathway and with restoration of NAD+ levels with NMN. Essentially, the scientists of this study want to find how disruptions of the NAD+ biosynthesis pathway affect muscle health along with neuromuscular junctions and whether NMN administration can ameliorate these defects.
Results of the study indicate cellular impairments in the mouse model with NAD+ biosynthesis impairments at the neuromuscular junction. To test effects of cellular impairments involving the NAD+ biosynthesis pathway, this team of scientists deletes a gene encoding the enzyme, Nampt. Nampt acts as the rate-limiting enzyme in the salvage pathway converting nicotinamide to NMN to facilitate NAD+ biosynthesis in mammals. Therefore, without the gene encoding Nampt protein, some disruption of NAD+ biosynthesis results. The scientists measure cellular trafficking of microscopic vacuoles containing signaling molecules (neurotransmitters) called ‘vesicles’ at the neuromuscular junction. The team measures release of the vesicles at the neuromuscular junction (exocytosis) along with formation and internalization of vesicles at the neuromuscular junction (endocytosis). The group finds impaired exocytosis and endocytosis of vesicles at the neuromuscular junction in mice without Nampt enzyme compared to mice with functional Nampt enzyme. Treating the mice without Nampt protein with NMN restores exocytosis and endocytosis as measured with fluorescence microscopy.
Lundt et al. (2020) investigates the structure of the neuromuscular junction with deletion of Nampt, using fluorescence microscopy. Compared to mice with intact Nampt enzyme, mice with Nampt enzyme deletion show significantly shorter neuromuscular junctions with smaller radii. When Nampt enzyme deletion mice receive NMN treatment, the scientists observe restorations of the neuromuscular junction structure.
The scientists find skeletal muscles have shorter relaxation periods following low electrical stimulation (low frequency) after Nampt enzyme deletion. In NMN-treated mice with Nampt enzyme deletion, a unique phenotype results, where muscles have diminished contractile force and also lose force more slowly. Thus, the scientists observe NMN administration produces an intermediate state between Nampt enzyme deletion and functional Nampt. The team obtains these results with examination of muscle contractions comparing mice with deleted Nampt enzyme to mice with functional Nampt enzyme. The team uses a method called patch-clamp analysis to analyze electrical forces (voltages) elicited from muscle contractions for this purpose.
Lundt et al. (2020) observes mice with Nampt enzyme deletion having microscopically misaligned muscles as measured with an electron microscope. Furthermore, treatment of mice having Nampt enzyme deletion with NMN restores alignment. In a similar experiment using electron microscopy, the group finds malformed mitochondria in skeletal muscle with Nampt enzyme deletion. The team of scientists observes NMN treatment cannot restore the mitochondria shape in skeletal muscle.
The results of the study present interesting information such as Nampt enzyme deletion facilitating structural problems and reduced vesicle cycling in the neuromuscular junction. Also interesting, muscle fiber structure and function has impairments with Nampt deletion. The scientists observe treatment with NMN can improve neuromuscular junction structure and vesicle cycling at the neuromuscular junction with Nampt enzyme deletion. The team also observes NMN treatment can ameliorate muscle misalignments but not muscle mitochondria malformations. According to the group, NMN administration can partially restore muscle function. As the authors note, the symptoms of mice with Nampt deletion resemble an ALS model. Moreover, NAMPT levels are decreased in the human ALS brain.2 Therefore, this study gives new insight regarding the NAMPT-mediated NAD+ salvage pathway in possible pathology of ALS. With the restorative properties of NMN demonstrated in the study, it suggests also NMN could provide a useful therapeutic agent for diseases of skeletal muscles like ALS.
 Comments
Comments