NMN Induces Metabolic Shift in Damaged Cells to Promote Survival
UC Irvine scientists demonstrate that damaged cells undergo a metabolic shift to survive that arises from NAD+ depletion.
Highlights
- Cell damage initiates a metabolic shift to promote survival
- Blocking the damaged cell metabolic pathway leads to cell death
- NMN restores damaged cell viability by reversing the metabolic shift
As we age, our cells and DNA accrue damage. To offset this, our cells have developed damage sensing responses with a battery of proteins. One such DNA damage sensing enzyme is called Poly(ADP-ribose) polymerase 1 (PARP1), which consumes the vital molecule nicotinamide adenine dinucleotide (NAD+) to repair DNA and promote cell survival. But the consequences of NAD+ consumption by PARP1 in DNA-damaged cells are unclear.
Digman and colleagues from UC Irvine published a study in Molecular Biology of the Cell that provided evidence for a novel metabolic response that promotes cell survival in response to damage. Their findings indicate cells undergo a metabolic shift when faced with PARP activation that involves switching pathways that cells use to generate energy.
“Our study provides an important paradigm for the mechanism of an inducible metabolic switch critical for cell survival against complex DNA damage,” stated the researchers in their publication.
These results clarify cell pathways related to aging and damage, which is critical for the therapeutic development of drugs called senolytics that clear aged, non-proliferating cells along with anti-cancer drugs.
Cells Can Produce Energy Through Two Pathways
Cells make energy from the breakdown of the sugar glucose through a process called glycolysis. They also muster energy from electron exchange reactions involving NAD+ and its electron-carrying form NADH in a mechanism called oxidative phosphorylation. Digman and colleagues found that PARP1 activation from experimentally-induced DNA damage not only triggered NAD+ depletion but also a shift in metabolic balance from glycolysis to oxidative phosphorylation. In other words, given a particular degree of cellular damage, cells begin relying more heavily on exchanging electrons to generate energy than breaking down sugars.
Cellular Damage Induces NAD+ Consumption and a Metabolic Shift
To characterize the metabolic state of cells in response to damage, the scientists measured glycolysis (extracellular acidification rate, or ECAR) and oxidative phosphorylation (oxygen consumption rate, or OCR) in damaged human cervical cancer cells (HeLa cells). The team induced damage to the cells with an agent called methyl methanesulfonate (MMS). This triggered the HeLa cells to more heavily rely on oxidative phosphorylation, as shown by a higher oxygen consumption rate to extracellular acidification rate ratio. Importantly, when the researchers inhibited PARP and supplemented the cells with nicotinamide mononucleotide (NMN), the strong metabolic shift to oxidative phosphorylation by the HeLa cells was reversed back to glycolysis. These findings provided evidence that DNA damage induces PARP activation, which diminishes NAD+ levels and triggers a metabolic shift toward reliance on oxidative phosphorylation.
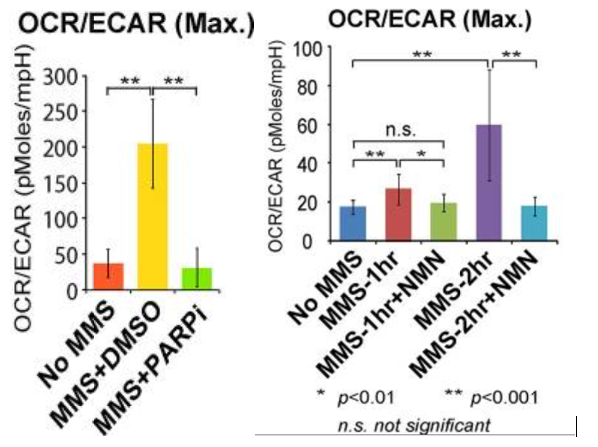
Damaged Cells Rely Heavily on Electron Exchange for Energy Production
To show how critical oxidative phosphorylation is for damaged cell energy production and survival, the researchers treated damaged cells with rotenone and antimycin, inhibitors of oxidative phosphorylation. Damaged cells treated with these inhibitors displayed major reductions in survival, but PARP inhibition was able to restore cell viability. This shows the importance of oxidative phosphorylation for damaged cell survival and how PARP1 activation mediates the metabolic shift toward reliance on oxidative phosphorylation.

Reliance on Electron Exchange for Energy Production Varies Between Cell Types
To determine whether the damage-induced metabolic shift mediated by PARP1 occurs only in human cancer HeLa cells, Digman and colleagues examined non-cancerous human cells — fibroblast cells HFF-1 and retinal cells ARPE-19. The cell damaging agent MMS produced significantly reduced ARPE-19 cell viability with reliance on PARP-mediated oxidative phosphorylation. On the other hand, they didn’t see these effects in the HFF-1 cells. They surmised that damage-induced oxidative phosphorylation reliance is not unique to HeLa cells but that there is significant cell type variability.

“We found that NAD+ consumption by PARP1 activation shifts the metabolic reliance to [oxidative phosphorylation], which is critical for damaged cell survival, uncovering a novel prosurvival metabolic response to DNA damage regulated by PARP1,” said the scientists.
Metabolic Pathway Insight May Provide Future Means to Treat Cancer and/or Rejuvenate Aged Tissue
The study provides a solution as to why many types of cancer cells must utilize oxidative phosphorylation for survival with high DNA damage levels and subsequent PARP activity. This may at least partially explain why cancer cells are more sensitive to oxidative phosphorylation inhibitors. Perhaps future studies can use these findings to come up with ways to induce cancer and aged, non-proliferating cell death for disease treatments and potential tissue rejuvenation therapy.
 Comments
Comments