Researchers Restore Cognition In Mice With Traumatic Brain Injury (TBI)
Study shows that NMN biosynthesis may have widespread clinical utility in the setting of neurodegenerative conditions
Highlights
- P7C3-A20 is an activator of the rate-limiting enzyme that produces the NAD+ precursor nicotinamide mononucleotide (NMN).
- Administration of P7C3-A20 in mice with traumatic brain injury (TBI) blocks chronic neurodegeneration and recovers normal cognitive function.
A major cause of the long-term disabilities that afflict survivors of traumatic brain injury (TBI) is chronic degeneration of the nervous system. This neurodegeneration is linked to an increased risk for late-life disorders, including Alzheimer’s disease, Parkinson’s disease, vascular dementia, and chronic traumatic encephalopathy. Despite the enormous societal burden of chronic neurodegeneration after TBI, there are currently no medicines that prevent or slow this disease process.
Vázquez-Rosa and colleagues from the Harrington Discovery Institute in Cleveland report in Proceedings of the National Academy of Sciences of the United States of America on the restoration of a semipermeable membrane separating blood from brain fluid — the blood brain barrier (BBB) — structure and function by a compound called P7C3-A20 when administered one year after TBI. This compound, which activates an enzyme called NAMPT that generates nicotinamide mononucleotide (NMN) in cells, stopped chronic neurodegeneration and recovery of normal cognitive function, benefits that persisted long after treatment cessation. These results may provide a new treatment option for patients who have suffered a remote TBI or other neurological conditions.
“Our results provide a rational foundation for developing treatments for patients suffering from chronic progressive neurodegeneration and cognitive dysfunction after remote TBI,” propose the researchers in their article.
The Bare Necessity of the Blood-Brain Barrier
For our brains to properly function, we rely on an extensive blood vessel network that works in concert with multiple cell types to form what’s called the neurovascular unit (NVU). The NVU rapidly responds to the changing metabolic needs of the brain. Through continuous crosstalk, the NVU forms an integrated system of neurovascular coupling that ensures the optimal supply of oxygen and micronutrients across the BBB consonant with the metabolic demand that varies with neuronal activity.
Also, the NVU protects the brain from exposure to injurious agents via the BBB, which prevents the entry of peripheral toxins while also mediating the removal of proteins and other substances from the brain. So, perhaps it is not surprising that disruption of the NVU and the BBB that it maintains can harm brain health, potentially leading to neurodegeneration.
Targeting NMN Generation to Protect the Brain
Nicotinamide adenine dinucleotide (NAD+) is a compound that has its hands in the function and survival of cells throughout the human body, and the brain and BBB are no exception. NAD+ has been shown to enhance the survival of brain cells and improve cognition in various models of nervous system disease and injury. Given the importance of NAD+ in brain health, the research community has sought ways to maximize NAD+ levels, either by boosting generation or inhibiting degeneration of the compound.
However, our cells don’t do a great job of processing a direct supply of NAD+. So, researchers have been trying to find ways to elevate NAD+ levels by making precursor molecules like NMN more accessible. NMN can be directly consumed by cells, and each molecule of NMN processed results in a molecule of NAD+. Cells can also be stimulated to generate NMN by activating an enzyme called nicotinamide phosphoribosyltransferase (NAMPT), which can provide a steady flow of the NAD+ precursor. For these reasons, there has been a recent push by researchers to test NAMPT activating compounds in preserving brain and BBB integrity during aging and disease.
P7C3-A20 Improves Cognitive Capacity after Traumatic Brain Injury
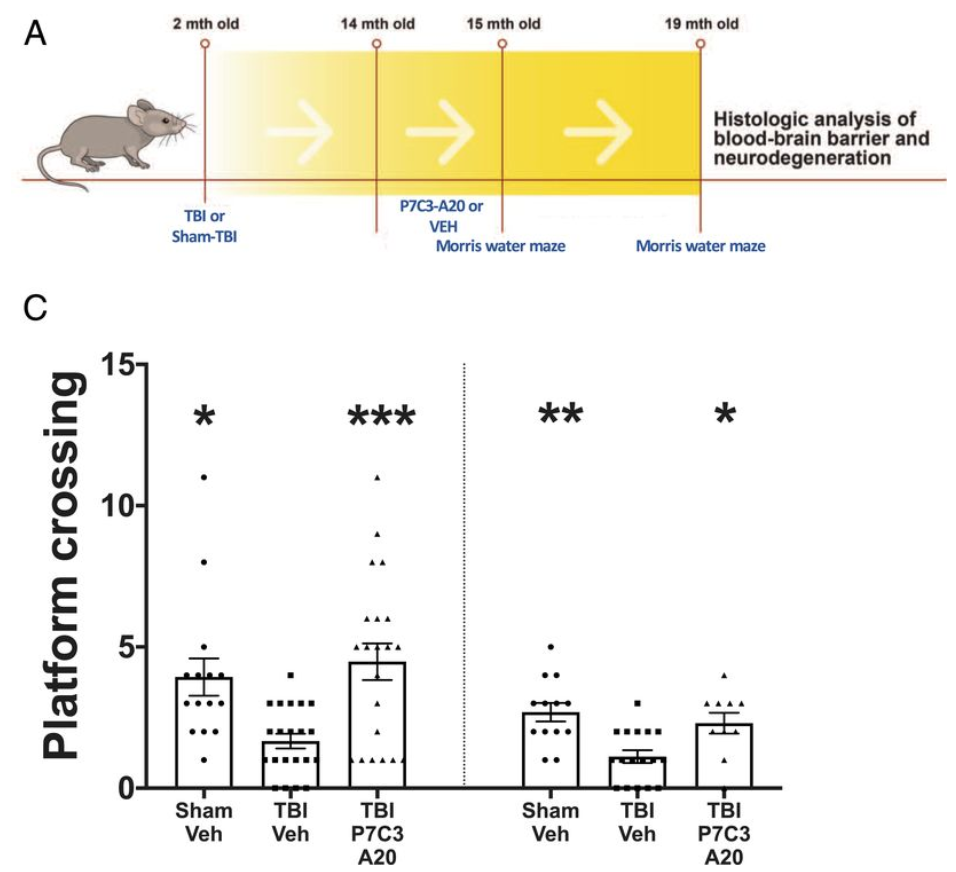
Here, Vázquez-Rosa and colleagues studied a mouse model of TBI that produces neurodegeneration and neurobehavioral deficits reminiscent of TBI in people. One year after mice experienced traumatic brain injury, these researchers treated the mice with the NMN generating compound P7C3-A20 for one month and then analyzed their brains and BBBs for structural and functional integrity. This late time point was selected because it represents approximately the midpoint of a typical mouse’s lifespan and thus can be considered a model of delaying initiation of treatment in people until decades after their TBI. This is important because of the great many people living today who are suffering from the chronic deficits of TBI. They then treated mice with the NAMPT activator P7C3-A20 beginning 1 year after a single injury.
Immediately after treatment completion, cognitive function in the 15-mo-old animals was evaluated through a spatial memory test called the Morris water maze. Although initial task learning was equivalent between groups, they observed significant memory deficits in the untreated mice with TBI while memory performance in the P7C3-A20 treated TBI mice was equivalent to the uninjured control animals. Four months later at 19 months of age (17 months post-injury), the untreated mice with TBI displayed impaired learning and memory whereas, once again, the TBI mice treated with the NAMPT activator again performed as well as the uninjured control animals.
Bolstering the Blood-Brain Barrier
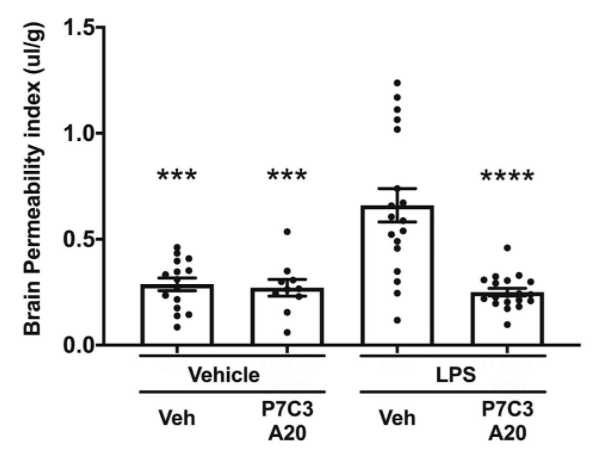
Vázquez-Rosa and colleagues show that deterioration of brain microvascular endothelial cells accompanies chronic axonal degeneration 1 year after TBI in mice. This is consistent with human studies of chronic TBI. For example, in one study, 47% of brains from long-term TBI survivors (up to 47 years after injury) showed histologic evidence of BBB deterioration. Recovery was achieved by 30 days of once-daily administration of the NAMPT activator P7C3-A20.
Four months after treatment with the NAMPT activator, microscopy revealed full repair of TBI-induced breaks in the BBB’s blood vessels. They also observed the restoration of normal BBB blood vessel length as well as several other indicators of improved BBB integrity. These changes were accompanied by cessation of TBI-induced chronic axonal degeneration. P7C3-A20 also protected mice from BBB degradation after acute TBI.
The Clinical Utility of NAMPT Activators
Taken together, these results suggest that BBB deterioration may be a major contributor to chronic neurodegeneration after remote TBI and that its repair may halt this pathology.
“At present, our results support the broad potential utility of a pharmacologic agent able to directly repair the BBB, a property that we demonstrate here is possessed by P7C3-A20,” concluded the authors.
Lastly, it is important to place into context how these results relate to the association between TBI and later neurodegenerative disease. BBB deterioration has also been observed in neurodegenerative conditions, including Alzheimer’s disease and other dementias, including Parkinson’s disease, vascular dementia, and chronic traumatic encephalopathy. These studies in mice might not apply to humans; however, if this compound passes toxicity studies in humans, its efficacy in protecting patients following TBI — and possibly in others at risk for these diseases — likely will be tested.
“Because TBI increases the risks of other forms of neurodegeneration involving BBB deterioration (e.g., Alzheimer’s disease, Parkinson’s disease, vascular dementia, chronic traumatic encephalopathy), P7C3-A20 may have widespread clinical utility in the setting of neurodegenerative conditions. We speculate that restoration of BBB integrity with P7C3-A20 after TBI could yield not only cognitive benefits but also reduce the otherwise increased risk for patients to develop other forms of neurodegenerative disease later in life,” concluded the authors.
 Comments
Comments