Study Demonstrates Anti-Aging Therapy for Pulmonary Fibrosis
Airway delivery of a longevity-linked gene resolves age-associated persistent lung fibrosis in mice
Highlights
- SIRT3 is decreased in the lungs of individuals with idiopathic pulmonary fibrosis and aged mice with lung injury.
- Airway delivery of Sirt3 DNA resolves chronic, age-associated lung fibrosis in aged mice.
Aging is linked to impaired regenerative capacity. So, it is not surprising that aging is an established risk factor for human fibrotic disorders, defined by the inability for tissues to heal, like the deadly and progressive age-related lung disorder called idiopathic pulmonary fibrosis. For these reasons, there is growing interest in how aging contributes to the risk and progression of age-related fibrosis, including idiopathic pulmonary fibrosis, and how to treat them.
Rehan and colleagues from the Tulane University School of Medicine published an article in Nature Aging implicating a critical role for the longevity-associated protein sirtuin 3 (SIRT3) in promoting resolution of lung fibrosis employing an aging mouse model. They found that SIRT3 levels were remarkably reduced in the lungs of humans with idiopathic pulmonary fibrosis, which was exacerbated in patients with rapid progression of the disease. Similarly, aged mice subject to lung injury failed to recover SIRT3 levels, which regained the capacity for fibrosis resolution with the restitution of SIRT3 levels.
“In this study, we report that SIRT3 is downregulated in fibroblasts from individuals with idiopathic pulmonary fibrosis and … in the lungs of aged mice with persistent, non-resolving fibrosis; restoring SIRT3 expression in the late reparative phase reverses established lung fibrosis,” concluded the authors. “We propose that aging biology can be leveraged to develop novel therapeutic strategies that target cellular plasticity and fate in established fibrosis.”
Regeneration and fibrosis: two sides of the same coin
Across organs in humans and many organisms, regeneration and fibrosis are juxtaposed. On the one hand, impaired regeneration leads to fibrosis, and on the other hand, a skewing of the tissue repair response to fibrosis may reciprocally dampen latent regenerative capacity.
These non-resolving fibrotic disorders may be related to the inability to ‘turn off’ the body’s repair response critical to the initial tissue injury. Along these lines, idiopathic pulmonary fibrosis is characterized by the persistent presence and activation of cells that are essential for wound healing called myofibroblasts that won’t go away or die off. So, these cells may be at the core of impaired regeneration of lung injury.
Longevity-linked genes SIRT3 and FOXO3A are key to age-related wound healing
Rehan and colleagues investigated a new role of the longevity-linked gene SIRT3 in promoting the resolution of lung fibrosis employing an aging mouse model. SIRT3 is part of the sirtuin family of enzymes that depend on nicotinamide adenine dinucleotide (NAD+) to function. They have different subcellular localizations and influence broad activities through target proteins that participate in cell survival and health. SIRT3 is localized to the power generating structure of the cell called mitochondria, and the loss of its activity is linked to accelerated aging, cancer, and age-related neurodegenerative diseases.
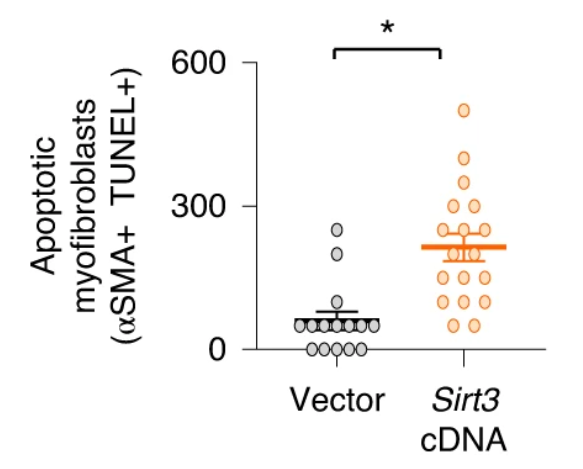
This research team reported a marked reduction of SIRT3 gene activation in the lungs of humans with idiopathic pulmonary fibrosis, which was further reduced in patients with rapid progression. This finding was mirrored in aged mice that failed to recover SIRT3 levels during the resolution phase of lung injury, which led to the formation of myofibroblasts that wouldn’t undergo cell death (apoptosis) and were locked in their cell cycle (senescence) and, ultimately, the development of lung fibrosis. But when Rehan and colleagues restored SIRT3 levels in the lung by airway gene delivery in aged mice, they saw that the rodents recovered their capacity for wound healing and the resolution of fibrosis as if they were young mice.
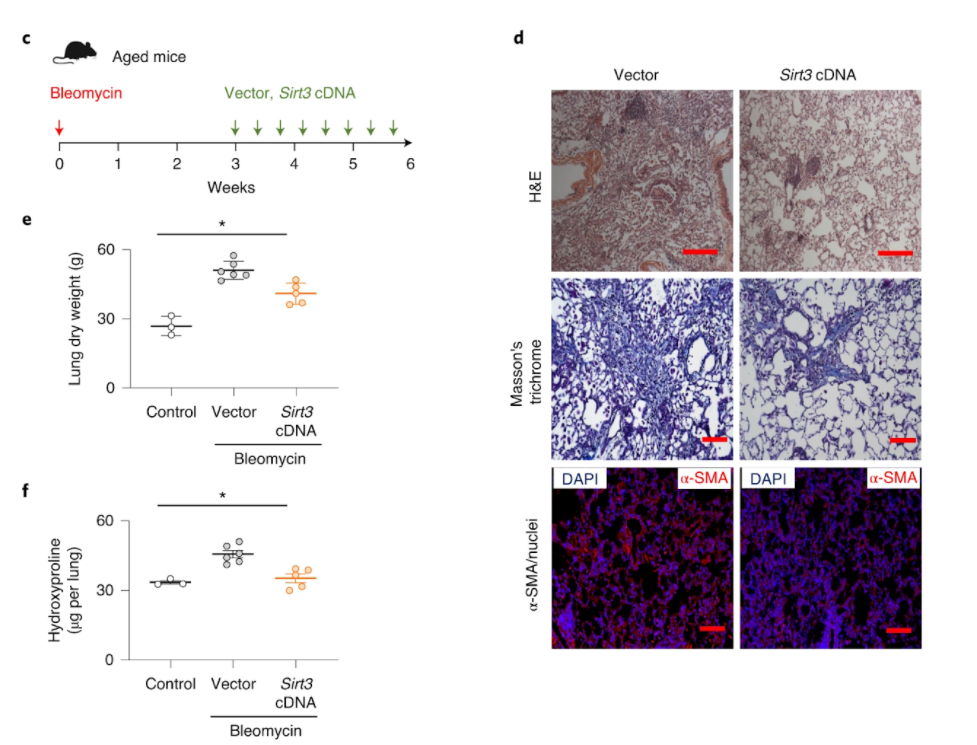
How SIRT3 mediates its anti-fibrotic effect appears to be related, at least partly, by directly modifying and activating FOXO3A in myofibroblasts, which recovered their susceptibility to programmed cell death or apoptosis. FOXO3A is a pro-longevity gene involved in many critical cellular processes including proliferation and apoptosis. Interestingly, when they excessively activated SIRT3 in immune cells involved in the injury response called macrophages, the protein FoxO3a was more efficiently activated in co-cultured fibroblasts.
Airway gene delivery of longevity-linked gene resolves lung fibrosis
So, it appears that SIRT3 is playing a critical role in modulating tissue regenerative capacity that involves some sort of cross-talk between macrophages and fibroblasts. These studies support a method where macrophages in the lung uptake the SIRT3 gene by airway delivery and produce factors critical to wound healing that can activate FoxO3a in myofibroblasts and mediate pro-apoptotic effects in these reparative cells.
“Together, our studies support a critical role for SIRT3 and FOXO3A in age-related stress responses and in cell-cell communication to orchestrate a pro-regenerative response to tissue injury,” concluded the authors. “Therapeutic strategies to restore these integrated longevity pathways to potentially reverse organ fibrosis deserve further study.”

 Comments
Comments