Several Anti-Aging Molecules Work in Animals, But Will They Work in Humans?
Although a growing number of studies show that It's possible to slow or even reverse aging, at least in mice, we're still waiting for major breakthroughs to prevent age-related diseases and increase lifespan.
Imagine a world with a cure for all chronic conditions from Alzheimer’s, cancer, and diabetes to aging. Well, that world exists — if you’re a laboratory mouse. When partaking in longevity-promoting experimentation, even some worms have lived a lifetime and a half longer than their non-tampered counterparts. Unfortunately for humans, a good chunk of these anti-aging and lifespan-prolonging approaches that work on animals don’t seem to translate to us. Many exciting biological discoveries related to altering the rate of aging in non-human species are sometimes not applicable or lost when we apply them to humans.
The move from slowing fundamental processes of aging in laboratory animals to slowing aging in humans will not be as simple as prescribing a pill and watching it work. Only after a therapeutic is confirmed as safe and effective in a preclinical model — like laboratory mice or human cells — can it be tested in people. Yet, most therapeutics don’t even make the hurdle of being safe in humans, let alone effective. A staggering 95% of drugs tested in patients fail to reach the market, despite all the promising animal studies that precede their use in humans. In the worst-case scenario, they’re deadly.
The TeGenero Incident and Other Clinical Trial Disasters
To illustrate, we can take the example of canines and chocolate. Humans are not genetically or physiologically different from dogs — our genetic compositions are roughly 84% the same; yet, we’re different enough that something we find to be a delightful dessert, like a Hershey’s bar, can be lethal for man’s best friend. That’s because dog livers are poor at breaking down a couple of chemicals found in chocolate (caffeine and theobromine). So, it doesn’t take much for toxic levels to build up in a dog’s bloodstream.
When you turn the tables on this concept, what happens is that humans can get put into dangerous situations during clinical testing; sometimes, they even die. One of the most famous examples is a clinical study conducted for an antibody called TGN1412 that preclinically demonstrated its therapeutic potential in autoimmune diseases, such as rheumatoid arthritis. In 2006, when researchers from TeGenero tested this antibody in six human volunteers, they had to immediately withdraw it from phase 1 clinical trials when six human volunteers faced life-threatening conditions involving multiorgan failure and were later moved to the intensive care unit.
In the case of TGN1412, at least part of the problem was that the drug’s target — a protein on specific immune cells — differs slightly between the monkey and human versions. The drug binds more strongly to the human immune cells and triggers a rapid release of massive amounts of chemicals involved in inflammation. Specific immune cells in mice lacked a receptor present in humans that strongly binds TGN1412, causing over-activation of the cells and the failure to predict a lethal ‘cytokine storm,’ where the immune system goes haywire, in humans.
The deadly outcome from the TGN1412 clinical study is not just a one-off event. In another clinical trial, an antiviral for Hepatitis B called Fialuridine showed adverse reactions in phase 2 clinical trials leading to the death of five human volunteers due to severe hepatic toxicity and lactic acidosis. Before being introduced into humans, Fialuridine was tested on different animals, including mice, rats, dogs, monkeys, and woodchucks. These studies demonstrated that doses hundreds of times higher than those administered to humans did not induce any toxic reactions. But none of the preclinical toxicity studies on laboratory animals could predict the toxic outcomes observed in phase II studies.
Fialuridine is toxic in humans because of a specific protein located on our mitochondria, the cell’s energy-generating structures. This protein transports the drug into the mitochondria, and once the drug is taken up, it poisons this essential cell component. This turns off the energy supply to our livers, where the drug is absorbed. Even though this protein is also present in mice, it does not send the medication into mitochondria because of only three differences in the DNA of mice. This trio of DNA mutations changes the gene encoding the protein just enough to keep it away from the cell’s mitochondria so the protein cannot transfer the drug into it.
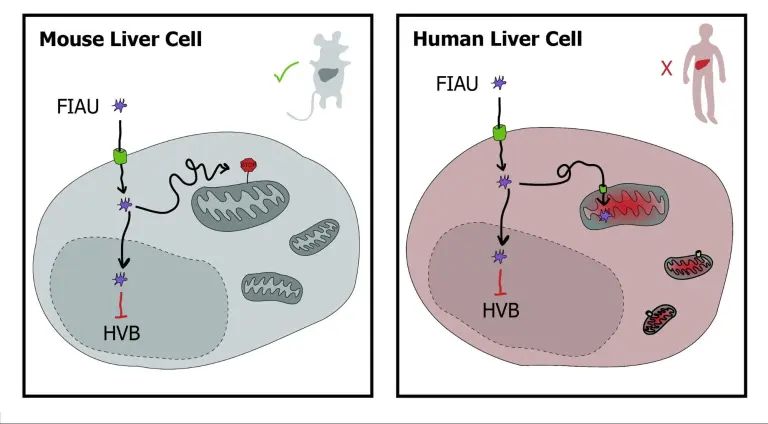
Though we share many genes with mice, they don’t necessarily do the same thing in both of us. The take-home message: though we may have the same genetic makeup, it doesn’t mean that the genes are activated in the same cells or play the same role in aging animals and humans.
The Human Element
And even if the same genes act in the same ways in the same cells to do the same things between lab animals and humans, there’s still the human variable. First of all, humans live much longer than rodents (about 30 to 40 times longer, on average). And since we’re not inbred like mice, we need many more subjects to account for different factors. So, anti-aging clinical trials aren’t exactly the cheapest ones around and require a lot of planning and patience.
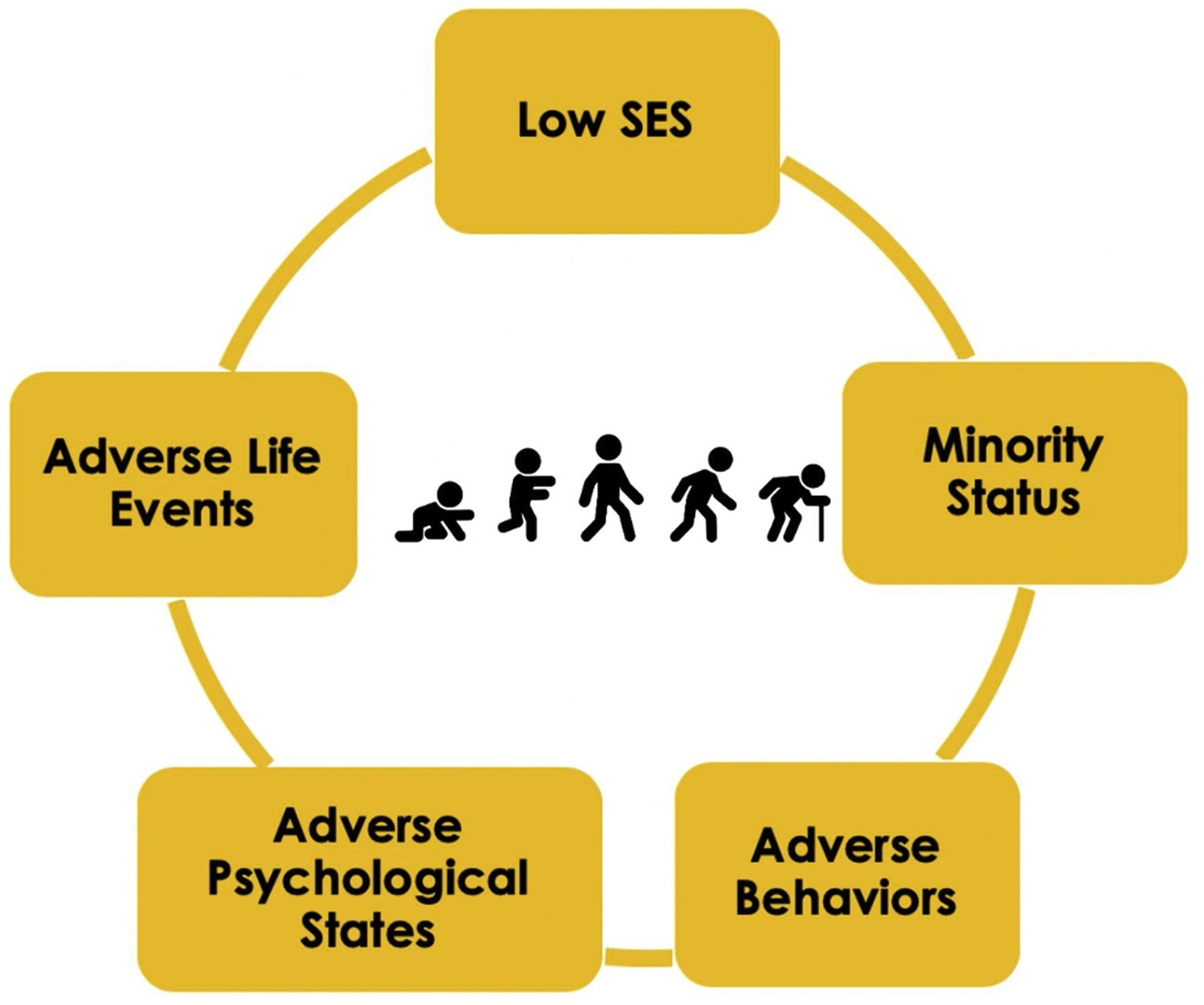
What’s more, compared to aging in laboratory animals and domestic pets, human aging has a greater variety of origins and influences, such as social and behavioral ones. These influences include potential intervention targets that are uniquely human and therefore are not easily investigated in animal research. Oft-studied behavioral and social influences on aging might include personality traits, intelligence, loneliness and social connection, purpose in life, stress, early-life adverse experiences, or even psychiatric history, which predicts early mortality.
Humans vary widely on such factors, which generates differences between individuals at the pace they age. Individual differences in the causes of peoples’ aging will complicate protein production from DNA by expanding alternative gene copies that mutes the effects of treatments, thus requiring extensive samples for aging science clinical trials.
A human adult’s pace of biological aging may be sped or slowed by genetics, which is far more complex than inbred lab mice, which are all essentially clones of one another. The early-life experiences and exposures and the differences in several behavioral and social lifestyle factors that do not characterize laboratory animals, like diet, physical activity, sleep, mental health, and smoking, have significant effects on healthspan and lifespan.
One study reported that individual differences in the pace of biological aging among adults tracked from age 26 to 38 were independently predicted by behavioral and social personal-history characteristics present in their childhoods. Adverse childhood experiences, social class, health, cognitive ability, and self-control, all measured in childhood, predicted differences between individuals in their pace of aging two decades later and did so over and above prediction from grandparents’ longevity. Participants who accumulated more of these psychosocial personal-history risks showed a faster pace of biological aging over the tracked decades.
Human-relevant factors like these have not been studied in animal models of aging. However, much human observational research shows that early-life behavioral and social risk factors can statistically predict strict aging outcomes, such as the timing of late-life disease onset and early mortality. There’s much more to be considered, including indicators of social status in childhood and adulthood, race and ethnicity, adverse experiences in childhood, adult trauma, negative psychological states, poor health behaviors, and age and gender. However, we don’t yet know how these social factors link to various age-related health outcomes, let alone how to account for them in clinical trials.
How to Avoid Getting Lost in Translation
By no means should we throw mice by the wayside in our search for anti-aging therapeutics that work in humans. Experiments in rodents have been essential in the development of therapeutics and treatment strategies from diabetes to cancer. For example, a gene therapy that worked to cure sickle cell disease in mice was recently successfully used to treat humans as well. That being said, we cannot solely depend on mice as being the barrier to entry on human testing. Even just studying human cells in isolation may not be enough. Some people suggest using non-human primates as an alternative, but these experiments are costly and, in some cases, don’t guarantee translation into humans (see FIAU).
Anti-aging clinical trials also need to be strategically designed. Researchers need to choose between the fundamental aging processes known as healthspan and resilience. Healthspan refers to the amount of time spent in good health, free of age-related diseases. Bouncing back to baseline functional and health status after an acute event is termed resilience. When we get older and are more likely to be frail, we’re less likely to be able to cope with major stressors such as surgery or heart attacks. Anti-aging molecules should be evaluated for their ability to extend healthspan and improve resilience.

Ultimately, there is no one-size-fits-all solution, and the best answer may not lie in the type of animal we use at all. In addition to optimizing the preclinical models, the best way to improve a drug’s success rate may be through accounting for the variation in human biology, both biological and social. Scientific advances will likely pick up steam when biological approaches incorporate some of the concepts in the social hallmarks of aging in experimental design to test pathways based on what has been shown to promote earlier aging in humans. Research needs to eventually involve human cohorts to measure complete life circumstances and complete biology to demonstrate the relative importance of proposed mechanisms and understand how to intervene in the aging process. Such work will set the stage for better interventions to improve health outcomes.
Which Anti-Aging Drugs Are Being Tested in Humans?
Metformin: this FDA-approved drug has been used successfully to treat diabetes for more than 60 years. Studies have already shown that metformin can delay aging in animals. Metformin is the focus of the Targeting Aging with Metformin (TAME) trial and the Metformin in Longevity Study (MILES). However, several clinical trials have already demonstrated that metformin negatively impacts muscle in response to resistance training in healthy older individuals, such as the Metformin to Augment Strength Training Effective Response in Seniors (MASTERS) study.
Rapamycin: this FDA-approved drug is labeled as an immunosuppressant and used to treat kidney transplant patients. AgelessRx, in affiliation with the University of California, is launching a formalized scientific study to evaluate the safety and effectiveness of Rapamycin in healthy adults for longevity. The Participatory Evaluation of Aging with Rapamycin for Longevity (PEARL) study is a double-blind, randomized, placebo-controlled trial. Rapamycin showed positive effects on skin aging when applied as a cream, but there is minimal information on the long term oral administration of rapamycin in humans.
NAD+ Precursors: various NAD+ precursor molecules, such as NMN, are actively being tested in clinical studies. Some clinical studies are still establishing the dosing and safety parameters for NAD+ precursors, while others have moved ahead to looking at the effect of these compounds on aging. For example, NMN is being evaluated as an anti-aging supplement in middle-aged and older (40-65 Years) adults. There have been several recent publications based on completed trials showing effects on several age-related conditions. Even the US military is testing out the effects of NMN on aging in soldiers.
Senolytics: these senescent cell targeting drugs range from being found naturally in plants (flavonoids), such as fisetin and quercetin, to being synthesized in a laboratory like dasatinib. There are currently several clinical trials being conducted on senolytics. Fisetin is being evaluated in the Alleviation by Fisetin of Frailty, Inflammation, and Related Measures in Older Adults (AFFIRM) trial, and dasatinib and quercetin in combination are being looked at in the Senolytic Therapy to Modulate the Progression of Alzheimer’s Disease (SToMP-AD) Study.
For now, we will have to do a bit of waiting to see if agents targeting basic aging processes in mice translate into interventions for major chronic diseases and age-related disabilities.
 Comments
Comments