A Therapeutic Approach to Falling NAD+ Levels After Brain Injury
Scientists elaborate how NAD+ depletion compromises the ability of mitochondria to counteract free radical and reactive oxygen species buildup.
Following brain injury and during the progression of neurodegenerative diseases, the major power-generating components of the cell, the mitochondria, release reactive molecules known as free radicals that can cause further cellular damage. Recently, scientists from the University of Maryland published a review in Brain Sciences where they elaborated on a possible molecular mechanism whereby mitochondria counteract the buildup of free radicals following brain injury and identified a potential therapeutic approach.
In their review, they posited that the breakdown–catabolism–of an essential molecule called nicotinamide adenine dinucleotide (NAD+) following brain injury reduces the capability of mitochondria to repair damage from free radical buildup, the accumulation of reactive molecules, in cells. The investigators suggest that boosting NAD+ levels with a molecule called nicotinamide mononucleotide (NMN) may reduce cellular damage from brain injury.
NAD+ Falls In Response to Brain Injury
NAD+ is essential for energy production in cells and has key roles in metabolic reactions. NAD+ also activates enzymes that play preventative roles in aging and disease by maintaining cellular health and correcting damage to the DNA molecules that encode our genomes. However, research shows that NAD+ levels fall with age in animals and humans and are also reduced in the mitochondria of damaged brain tissue.
The balance between the production of reactive oxygen species–unstable molecules that contain oxygen–and the efficiency of the cellular detoxification process determines the levels of harmful reactive oxygen species. Generation of reactive oxygen species and a cellular process called free radical detoxification, whereby enzymes protect cells against free radical damage, depend on NAD+ levels. Reduced levels of NAD+ in mitochondria increase the production of reactive oxygen species like superoxide and inhibit free radical detoxification, such as superoxide detoxification.
Reduced levels of NAD+ inhibit free radical detoxification by diminishing the effectiveness of certain enzymes. One enzyme that is dependent on NAD+ to function is called sirtuin 3 (SIRT3), which removes molecular tags that inhibit the function of proteins through a process called deacetylation. When sufficient NAD+ levels facilitate the function of SIRT3, it deacetylates and, thus, activates an enzyme called mitochondrial superoxide dismutase (MnSOD), which is an essential mitochondrial antioxidant enzyme that detoxifies reactive oxygen species.
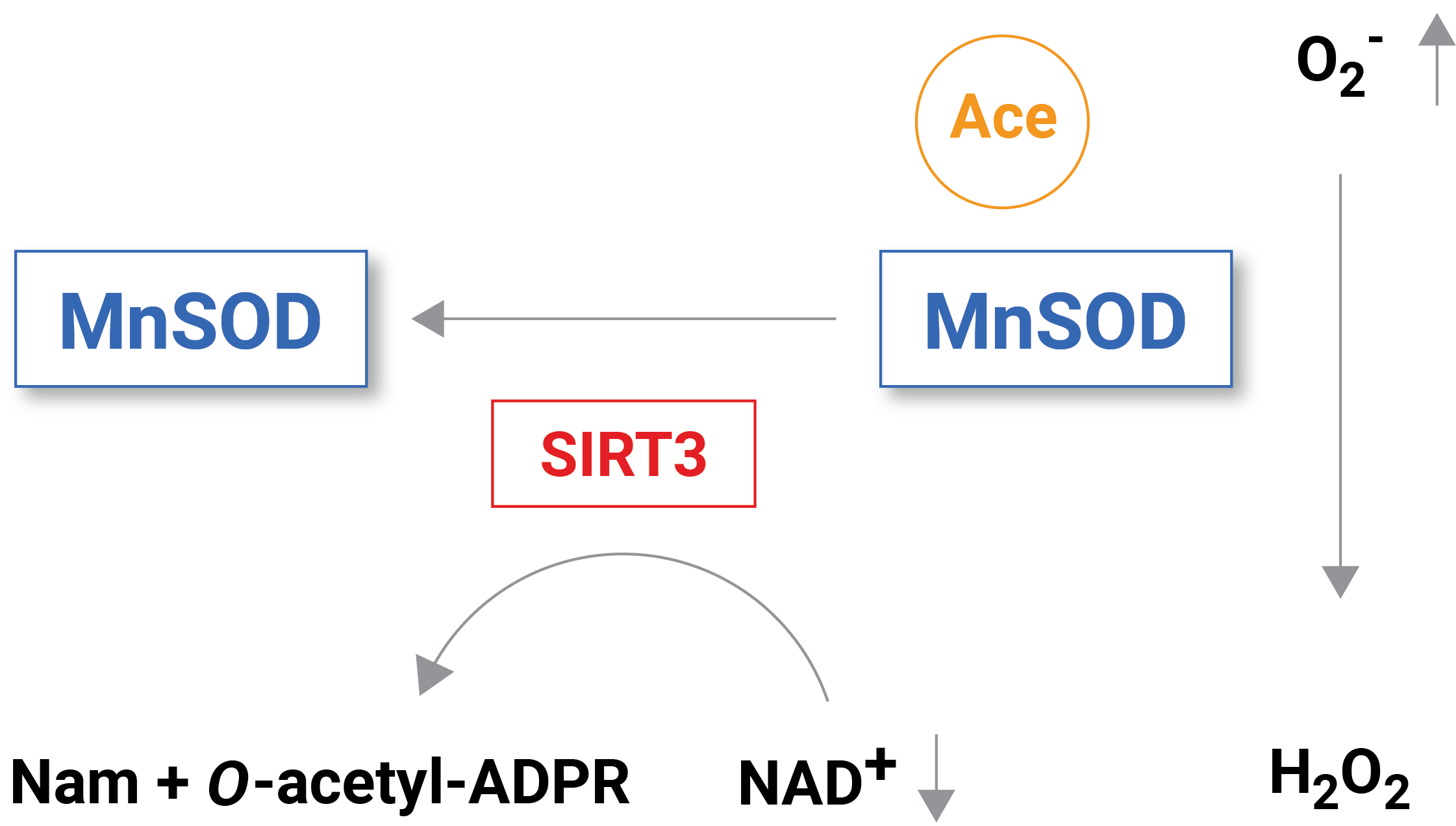
(Klimova et al., 2020 | Brain Sciences) Reduced NAD+ levels following brain injury inhibit the activity of the NAD+ dependent enzyme SIRT3. MnSOD acetylation accumulates without SIRT3 to remove acetylation (i.e., deacetylation). With inhibited MnSOD activity, the reactive oxygen species superoxide (O2–) builds up causing mitochondrial pathology.
When NAD+ levels are reduced in situations like brain injury, enzymes with essential functional roles in reactive oxygen species detoxification like MnSOD are inhibited, leading to free radical and reactive oxygen species buildup. “Our published data show that the delayed increase in brain tissue ROS levels following an acute brain injury is due to inhibition of the mitochondrial antioxidant mechanisms triggered by mitochondrial NAD+ catabolism and consequent hyperacetylation of enzymes, whose activities are essential for superoxide detoxification,” stated the investigators in the paper.
Restoring NAD+ With NMN
Researchers have used a molecule to boost NAD+ levels called nicotinamide mononucleotide (NMN) to counteract the age-related decline in reduced cellular NAD+ levels with age. The authors of this review suggest that NMN supplementation may constitute a therapeutic approach to mitigate the effects of reduced NAD+ levels from brain injury by preventing the depletion of NAD+ levels in mitochondria. Since increased concentrations of reactive oxygen species in mitochondria are damaging to cells, using NMN may improve clinical outcomes following reduced blood flow to the brain from injury.
 Comments
Comments