Why NAD+ Declines With Age
Long and healthy lifespans require NAD+, so why do levels of this vital molecule drop as we age?
From the simplest single-celled organisms to humans, all of life cannot exist without a molecule called nicotinamide adenine dinucleotide (NAD+). This molecule has its hands in many processes necessary for humans — and many organisms examined like yeast, flies, worms, and mice — to live a long and healthy life, let alone just being alive. NAD+ plays a role in all sorts of cellular tasks, including basic yet essential functions like helping electrons bounce around between other molecules to carry out many of the cell’s necessary chemical reactions as well as sensing and regulating metabolism.
Simply put, the available pool of NAD+ in a cell is governed by its generation and consumption. But our supply of NAD+ is not immutable; it doesn’t get perfectly recycled. We need to constantly supply ourselves, typically through our diets, with sources for NAD+. Yet as we age, NAD+ levels appear to drop like a runaway train that can never be caught even if all else, such as our diet, stays the same.
The cost of NAD+ decline comes at a severe price. The decline in NAD+ levels appears to play a crucial role in the development of metabolic dysfunction and age-related diseases. This is why there is a growing emphasis on figuring out ways to boost NAD+ levels.
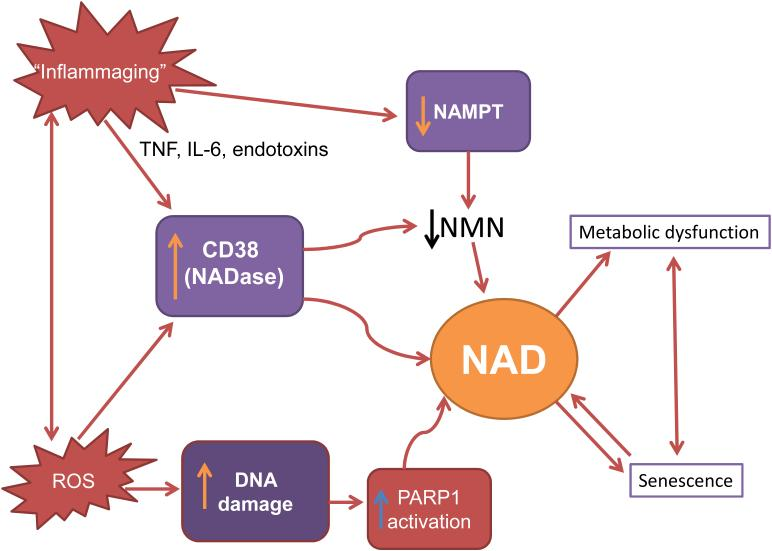
To optimally intervene in the downward spiral of NAD+ levels, we must understand the mechanisms that lead to cellular NAD+ decrease during aging, particularly whether the decline is mediated primarily by changes in its degradation synthesis, or both. Furthermore, it is critical to understand how specific cellular processes like DNA damage, inflammation, and senescence — the age-related freezing of cell replication — impact cellular NAD+ metabolism during the aging process.
Impaired NAD+ biosynthesis in aging
One reason that NAD+ levels drop with age is that it simply isn’t produced at the same rates from the precursor molecules available. For reasons yet unclear, the levels of NAMPT — the enzyme that generates the majority of NAD+ — diminish with age and, in turn, affect the activities of proteins dependent on NAD+. Exactly how NAMPT levels decline with age appears to depend on several factors.
Alterations to our body’s clock, or what’s known as the circadian rhythm, have been shown to contribute to age-associated NAMPT decline. Aging affects both the pace and strength of the circadian rhythm. A poorly timed or weak circadian rhythm drives the insufficient generation of NAMPT, which in turn will lead to decreases in NAD+ levels.
Another possible mechanism of age-related NAMPT decline is chronic inflammation. The damage caused by internal and environmental stressors that promote chronic inflammation accumulate with aging in different metabolic tissues including fat, skeletal muscle, and liver. In affected tissues, inflammatory signaling molecules called cytokines that can exacerbate cellular damage are released. These cytokines are reported to decrease NAMPT levels.
Inflammatory cytokines and oxidative stress appear to be major contributors to the development of chronic inflammation during aging. Since both inflammatory cytokines and oxidative stress contribute to the development of chronic inflammation during aging, chronic inflammation could be a reason by which both NAMPT-mediated NAD+ biosynthesis and circadian machinery are compromised during aging.
NAD+ degradation as a cause of age-related decline
There are many enzymes that consume NAD+ known to mediate a plethora of fundamental cellular processes. Three major enzymes are the PARPs, CD38, and SARM1.
PARPs are major NAD+ consumers. Among many PARP family members, PARP1 and PARP2 are major NAD+ consumers, responding to DNA strand breaks and facilitating the DNA repair process. During aging, PARP activation, possibly due to constant DNA damage from internal and external stressors like oxidative stress and UV-radiation, respectively, appears to contribute to significant decreases in intracellular NAD+.
CD38 is one of the primary enzymes that consume NAD+ in mammals and can modulate the NAD+ levels. Studies have shown that CD38 protein levels increase in multiple tissues and organs over age, contributing to NAD+ decline. Also, CD38 levels and activity are induced by cytokines and bacterial toxins. So, the chronic inflammation observed during aging could lead to an increase in expression of CD38 and subsequently cause NAD+ decline.
The use of NAD+ by the enzyme SARM1 is central to the degeneration of axons after injury and in the initiation phases of several neurodegenerative diseases. NAD+ depletion mediated by SARM1 leads to the degeneration of damaged neurons, a key event in early stages of age-related neuronal disorders, such as Parkinson’s disease, Alzheimer’s disease, and Amyotrophic Lateral Sclerosis (ALS).
Sirtuins and the consequences of the age-related NAD+ decline
The combination of decreased biosynthesis and increased breakdown of NAD+ exacerbates its depletion, causing a variety of age-associated conditions and diseases. Which one — an insufficient generation or overstimulated consumption — contributes further to the shortage of NAD+ may depend on cell types and tissues. No matter what causes NAD+ decline, it seems that major downstream mediators are sirtuins.
In humans, the sirtuin family is composed of seven members (SIRT1-SIRT7), all of which play a role in several cellular metabolic processes related to healthspan and longevity. Generally, sirtuins remove a modification called acetylation from target proteins to regulate reactions associated with vital processes such as metabolism, DNA and cellular repair, stress responses, circadian rhythm, and other cellular processes. By mediating such broad functions, sirtuins are evolutionarily conserved regulators for aging and longevity in diverse organisms.
These various NAD+-dependent functions of sirtuins place them in a key position for regulating aging and longevity in diverse organisms. For example, mice that generate more SIRT1 than typical brains see delays in aging and extended lifespans. Similarly, SIRT1 activation has therapeutic potential in neurodegenerative diseases like Huntington’s disease and amyotrophic lateral sclerosis (ALS). Also, mice that generate more SIRT6 throughout the body show lifespan extension.
The NAD+ downward spiral
NAD+ biosynthesis, mainly mediated by NAMPT and NAD+ consumption by NAD+ consuming enzymes, are in a delicate balance so that perturbations to either side can cause significant derailment of the system. A vicious cycle may exist in which molecular mechanisms involved in the aging process, such as oxidative stress, DNA damage, senescence, and inflammation, lead to tissue NAD+ decline, which subsequently exacerbates the processes that caused its decline in the first place.
As NAD+ is a common substrate between PARPs and SIRT1, there is a competition between their activities. If NAMPT-mediated NAD+ biosynthesis is disturbed or if NAD+ consumption is increased because of chronic DNA damage that elicits PARP activation, the intracellular NAD+ pool is decreased, and SIRT1 activity is reduced, causing an organism’s functional decline.
For example, PARP is chronically activated in aging worms and mice (liver or skeletal muscle). A possible explanation for these findings is that aging is associated with an increase in chronic nuclear DNA damage, which leads to NAD+ depletion by PARP. Since the loss of SIRT1 or SIRT6 activity exacerbates DNA damage may create an autocatalytic downward spiral in the nucleus with NAD+ deficit as the nexus.
Deleting PARP1 and PARP2 enhances SIRT1 activity, resulting in increased mitochondrial content, metabolism, and protection from diet-induced obesity. In mice lacking PARP1, there was a systemic elevation in NAD+ levels, SIRT1 activity, and metabolic benefits in these mice. Notably, PARP inhibitors boost NAD+ levels and increase SIRT1 activity to restore mitochondrial fitness and function.
Parallel findings were also reported for mice with the elimination of another NAD+-consuming enzyme, CD38, as shown previously. CD38-dependent modulation of NAD+ can alter the activity of SIRT1 and other sirtuins, as well as other NAD+-consuming enzymes, and affect cellular signaling and metabolism. These studies show clearly that PARP, CD38, and the nuclear sirtuins all compete for the same pool of NAD+, and inhibition of PARP or CD38 has the potential of activating sirtuins.
To close the loop, there is some evidence indicating that levels of NAMPT decline during replicative senescence of human smooth muscle cells and in peripheral tissue of old mice, such as white adipose tissue and skeletal muscle. Senescence also activates the expression of CD38 in tissue-resident macrophages, and proinflammatory signaling molecules released from senescent cells increase the activity and the expression level of CD38.
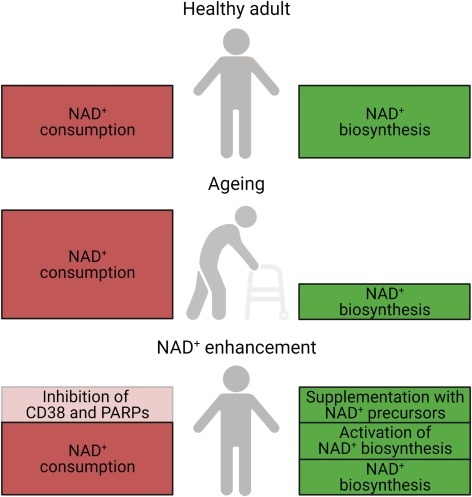
How to stop the NAD+ drain
Strategies to suppress chronic inflammation and sustain NAD+ biosynthesis and circadian function with aging might be effective in maintaining sirtuin activity and possibly robust health. Along these lines, various approaches to address the imbalance in NAD+ synthesis and consumption have been established.
There has been a lot of work examining the potential NAD+ precursor supplementation. There are five major precursors and intermediates to synthesize NAD+: tryptophan, nicotinamide, nicotinic acid, nicotinamide riboside (NR), and nicotinamide mononucleotide (NMN).
In addition, to increase NAD+ levels, activation of the rate-limiting enzyme, NAMPT, has been explored. Small molecule NAMPT activators like SBI-797812 and P7C3 are a pioneering approach to raise intracellular NAD+ and realize its associated salutary effects. NAD+ biosynthesis can be stimulated by the inhibition of an enzyme called ACMSD. Genetic and pharmacological inhibition of ACMSD boosts NAD+ synthesis and SIRT1 activity, ultimately enhancing mitochondrial function.
Alternatively, NAD+ consumption can be lowered by inhibition of CD38 and PARPs. Several CD38 inhibitors have been discovered, including the naturally occurring apigenin and the small molecule 78c. Olaparib, sold under the brand name Lynparza, is a PARP inhibitor used as a medication for the maintenance treatment of BRCA-mutated advanced ovarian cancer in adults. DSRM-3716 was recently identified as a potent inhibitor of SARM1’s NAD+ consuming activity.
In conclusion, NAD+ metabolism is very complex and includes several pathways and metabolites that need to be integrated into our understanding of the aging process. Future research will determine how mechanisms involved in aging, such as oxidative stress, DNA damage, inflammation, and cellular senescence, regulate and are regulated by NAD+ metabolism. In addition, it is crucial to determine the specific enzymes involved in the age-related NAD+ decline.
Despite the considerable advances that have been made in the past decade, several questions remain unanswered. Perhaps, among the most pressing questions, the molecular mechanisms and pathways linking NAD+ and aging continue to be elusive and not fully understood.
 Comments
Comments